Приложение 13
________________
* Для обеспечения идентичности текста с Правилами GMP ЕС в нем сохранены ссылки на нормативно-правовые документы ЕС.
Общие положения
Лекарственные средства для клинических исследований должны быть изготовлены в соответствии с правилами производства лекарственных средств (GMP), изложенными в настоящем стандарте.
Следует принимать во внимание и другие руководства с учетом стадии разработки препарата. Методики работы должны быть гибкими и допускать внесение изменений по мере получения дополнительных данных о процессе и соответствовать стадии разработки препарата.
При клинических исследованиях может возникнуть дополнительный риск для субъектов исследований по сравнению с пациентами, которые принимают имеющиеся на рынке препараты. Целью правил GMP в применении к производству лекарственных средств для клинических исследований является исключение риска для субъектов исследований и влияния на результаты клинических исследований недостаточных безопасности, качества или эффективности, обусловленных недостатками в производстве. В равной мере эти требования предназначены для обеспечения постоянства от серии к серии одного и того же лекарственного средства для исследований, используемого в одном или в разных клинических исследованиях, а также документального оформления и обоснования изменений в процессе разработки исследуемого лекарственного средства.
Производство лекарственных средств для исследований связано с дополнительной сложностью по сравнению с зарегистрированными лекарственными средствами из-за отсутствия установленного порядка, разных схем клинических исследований и, как следствие, разных требований к упаковкам, необходимости рандомизации и кодирования (маскирования, использования слепого метода), а также из-за большого риска перекрестных загрязнений и перепутывания препаратов. Более того, данные об эффективности и токсичности препарата могут быть неполными, процесс может быть не аттестован в полном объеме или могут использоваться зарегистрированные лекарственные средства, которые были переупакованы или модифицированы.
В связи с этим персонал должен в полной мере понимать правила GMP в отношении лекарственных средств для исследований и пройти соответствующую подготовку. Должно быть установлено взаимодействие со спонсорами клинических исследований, на которых лежит вся ответственность по вопросам клинических исследований, включая качество лекарственных средств для исследований.
Возросшая сложность технологических процессов требует применения высокоэффективной системы обеспечения качества.
В данном приложении также установлены требования к оформлению заказов, отгрузке, транспортированию и возврату материалов для клинических исследований, которые взаимосвязаны и дополняют руководство по клиническим исследованиям (Good Clinical Practice).
Примечание - Субъектам клинических исследований могут быть предоставлены препараты, которые не являются испытуемым препаратом, плацебо или препаратом сравнения. Такие препараты могут применяться для сопутствующей терапии или для оказания медицинской помощи с целью профилактики, диагностики или лечения, и/или вследствие необходимости обеспечения адекватного медицинского ухода, предусмотренного для субъектов исследований. Такие средства также допускается применять в установленном порядке для того, чтобы вызвать физиологическую реакцию. Эти препараты не являются лекарственными средствами для исследований и могут быть предоставлены спонсором или исследователем. Спонсор должен гарантировать, что данные препараты соответствуют запросу (заявке) на разрешение проведения клинических исследований и имеют требуемое качество с учетом цели исследований. При этом он должен принять во внимание источник препаратов, являются ли они зарегистрированными лекарственными средствами и были ли они переупакованы. Рекомендуется привлекать к этой работе уполномоченное лицо и учитывать его мнение.
Термины и определения
слепой метод, кодирование, маскирование (blinding): Процедура, при которой одна или более сторон, участвующих в исследовании, остаются в неведении относительно цели(ей) данного исследования. Единичный слепой метод означает неосведомленность субъектов исследования, а двойной слепой метод - неосведомленность о целях клинического исследования субъектов исследования, исследователей, наблюдателей и, в некоторых случаях, лиц, анализирующих полученные данные. В отношении лекарственного средства для клинических исследований слепой метод означает преднамеренное маскирование идентичности этого лекарственного средства в соответствии с указаниями спонсора. Декодирование (снятие маскировки) означает раскрытие информации об идентичности препарата.
клиническое исследование (clinical trial): Исследование, проводимое с участием человека в качестве субъекта для выявления или подтверждения клинических, фармакологических и/или других фармакодинамических эффектов исследуемого лекарственного средства, и/или нежелательных реакций на него, и/или изучения его всасывания, распределения, метаболизма и выведения с целью оценки его безопасности и/или эффективности.
препарат сравнения (comparator product): Разрабатываемое или имеющееся на рынке лекарственное средство (для положительного контроля) или плацебо, используемое в качестве препарата сравнения при проведении клинических исследований.
лекарственное средство для клинических исследований (investigational product): Дозированная лекарственная форма активной фармацевтической субстанции или плацебо, исследуемая или используемая в качестве препарата сравнения при проведении клинического исследования, включая уже зарегистрированные лекарственные средства, если способ их применения или производства (лекарственная форма или упаковка) отличается от зарегистрированной формы, или в случае их использования по еще не одобренным показаниям, или для получения дополнительной информации об уже зарегистрированной дозированной лекарственной форме.
первичная упаковка (immediate packaging): Контейнер или другой вид упаковки, имеющий непосредственный контакт с лекарственным средством или исследуемым лекарственным средством.
исследователь (investigator): Лицо, ответственное за проведение клинического исследования в месте его выполнения. При проведении клинического исследования группой лиц исследователем является руководитель группы, который может называться ведущим исследователем.
производитель/импортер лекарственного средства для клинических исследований (manufacturer/importer of investigational medicinal products): Лицо, имеющее право на производство (импорт) исследуемого лекарственного средства, оформленное в установленном порядке.
заказ (order): Задание на приготовление, упаковку и/или доставку определенного количества единиц лекарственного средства для клинических исследований.
наружная упаковка (outer packaging): Упаковка, в которую помещена первичная упаковка.
досье на лекарственное средство (product specification file): Комплект документов, содержащих всю информацию, необходимую для составления подробных инструкций по изготовлению, упаковке, контролю качества, выдаче разрешения на реализацию серии и отгрузке лекарственного средства для клинических исследований.
рандомизация (randomization): Присвоение субъектам исследований или контрольным группам обозначений, содержащих элемент случайности, с целью снижения возможности необъективного заключения.
код рандомизации (randomization code): Перечень, содержащий описание лечения каждого субъекта исследований с учетом рандомизации.
транспортирование (shipping/dispatch): Операции по упаковке и транспортированию заказанных лекарственных средств для клинических исследований.
спонсор (sponsor): Физическое или юридическое лицо, являющееся инициатором клинического исследования и несущее ответственность за его организацию и финансирование.
Обеспечение качества
1 Система обеспечения качества, разработанная и проверенная производителем или импортером, должна соответствовать требованиям настоящего стандарта, относящимся к лекарственным средствам для исследований, должна быть документально оформлена и быть доступна спонсору клинического исследования.
2 Спецификации и технологические инструкции на лекарственные средства для клинических исследований могут изменяться в процессе их разработки. В связи с этим должен быть организован их контроль и прослеживаемость всех изменений.
Персонал
3 Весь персонал, имеющий отношение к лекарственным средствам для клинических исследований, должен пройти подготовку, связанную со спецификой данного вида продукции.
4 Уполномоченное лицо несет особую ответственность за то, чтобы выполнялись все требования данного приложения. Для этого уполномоченное лицо должно иметь хорошую подготовку в области разработки препаратов, процессов и клинических исследований. Руководство для уполномоченного лица по оценке (сертификации) лекарственных средств для клинических исследований приведено в пунктах 38-41 настоящего приложения.
Помещения и оборудование
5 При работе с лекарственными средствами для клинических исследований информация о токсичности, эффективности и сенсибилизирующей способности может быть неполной, в связи с чем следует уделить особое внимание сведению к минимуму риска перекрестных загрязнений. Конструкция оборудования и проект помещений, методы испытаний и контроля и допустимые концентрации остатков после очистки должны учитывать природу этих рисков. Следует обратить внимание на организацию работы циклами (кампаниями), если это целесообразно. При выборе моющего средства следует учесть растворимость в нем препарата.
Документация
Спецификации и инструкции
6 Спецификации (на исходные материалы, первичные упаковочные материалы, промежуточные продукты, нерасфасованную и готовую продукцию), промышленные регламенты, технологические инструкции, а также инструкции по упаковке продукции должны быть, по возможности, полными для существующего уровня знаний. По мере разработки препарата их следует периодически пересматривать (при необходимости). Каждая новая версия документа должна учитывать последние данные, используемую технологию, нормативные и фармакопейные требования со ссылкой на предыдущую версию для обеспечения прослеживаемости. Любые изменения, которые могут иметь последствия для качества препарата, в частности для его стабильности и биоэквивалентности, следует вносить в соответствии с письменными требованиями.
7 Обоснования для изменений следует оформлять документально. Последствия изменений для качества препарата и последующих клинических испытаний необходимо анализировать и оформлять документально.
Заказ
8 Заказ должен содержать требование на изготовление и/или упаковку определенного числа единиц продукции и/или ее отгрузку. Заказ производителю дается спонсором или лицом, действующим по его поручению. Заказ должен быть оформлен в письменном виде (но может передаваться и электронным способом) и быть достаточно четким во избежание разночтений. Заказ должен быть официально утвержден и иметь ссылку на утвержденное досье на лекарственное средство или протокол клинических испытаний.
Досье на лекарственное средство
9 Досье на лекарственное средство должно непрерывно обновляться по мере разработки препарата, обеспечивая логическую связь с предыдущими версиями. Досье должно включать в себя следующие документы (или содержать на них ссылки):
- спецификации и аналитические методики для исходных и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции;
- технологические процессы;
- методики внутрипроизводственного контроля;
- утвержденную копию этикетки;
- протоколы клинических исследований и коды реализации (при необходимости);
- технические соглашения с заказчиками по контрактам (при необходимости);
- данные о стабильности;
- условия хранения и транспортирования.
Данный перечень не является исчерпывающим. Он может изменяться в зависимости от препарата и стадии его разработки. Содержащаяся в нем информация должна служить основой при оценке готовности для приемки и выдачи разрешения на выпуск уполномоченным лицом, которое должно иметь доступ к этой информации. Если различные производственные стадии проводят в разных местах, ответственность за которые несут разные уполномоченные лица, допускается вести отдельные досье с ограниченной информацией, относящиеся к определенному месту.
Промышленные регламенты и технологические инструкции
10 Каждая производственная операция или операция по отгрузке должна выполняться в соответствие с четкой и достаточно полной письменной инструкцией и сопровождаться оформлением протокола. Если операция не является повторяемой, то не обязательно составлять промышленный регламент и технологические инструкции. Протоколы имеют особое значение для подготовки окончательных текстов документов, которые будут использоваться при серийном производстве после получения регистрационного удостоверения.
11 Информацию, содержащуюся в досье, следует использовать при разработке подробных инструкций по изготовлению, упаковке, испытаниям для контроля качества, условиям хранения и транспортирования.
Инструкции по упаковке
12 Лекарственные средства для клинических исследований должны упаковываться индивидуально для каждого субъекта испытаний. Количество единиц упаковываемой продукции должно быть определено до начала операций по упаковке с учетом количества единиц, необходимых для проведения контроля качества, и архивных образцов. После окончания упаковки и маркировки необходимо подвести баланс (выполнить сопоставление) каждого вида упаковочных материалов, нерасфасованной и готовой продукции для каждой стадии производства.
Протоколы производства, испытаний и упаковки серии продукции
13 Протоколы производства и упаковки серии продукции должны содержать подробную информацию, достаточную для прослеживания всей последовательности операций, а также все существенные замечания, дающие новую информацию о продукте и внесении изменений, позволяющих получить дополнительные данные о продукции, усовершенствовать производственный процесс и технологические инструкции.
14 Протоколы производства серий продукции должны храниться не менее пяти лет после завершения или официального прекращения последнего клинического испытания, в котором была использована эта серия*.
________________
* Эти требования установлены в ст.9 Директивы 2003/94/ЕС.
Производство
Упаковочные материалы
15 В спецификациях и инструкциях (методиках) контроля качества должны быть указаны меры по предотвращению случайного снятия кодирования (маскировки) из-за различий внешнего вида разных серий упаковочных материалов.
Технологические операции
16 На стадии разработки лекарственного средства следует определить критические параметры и виды внутрипроизводственного контроля для контроля технологического процесса. Временные параметры и виды внутрипроизводственного контроля могут быть получены из прошлого опыта, в том числе, из предыдущих исследований. Руководящий персонал должен уделять особое внимание разработке инструкций и постоянному их развитию с учетом опыта, приобретаемого в процессе производства. Контролируемые параметры должны быть обоснованы в соответствии с имеющейся в данное время информацией.
17 Не обязательно проводить аттестацию (испытания) технологических процессов производства лекарственных средств для исследований в объеме, предусматриваемом для серийного производства, но помещения и оборудование должны быть аттестованы. Для стерильных лекарственных средств аттестация (испытания) процессов стерилизации должна проводиться по тем же стандартам, что и для серийного производства продукции, разрешенной к реализации.
Аналогичным образом (при необходимости) следует подтверждать эффективность инактивации/удаления вирусов и/или других загрязнений биологической природы для обеспечения безопасности продукции биотехнологического производства в соответствии с принципами и методами, содержащимися в руководствах в данной области.
18 Аттестация (испытания) асептических процессов представляет особую трудность при малых размерах серий продукции. В этих случаях число единиц, наполняемых средами, может быть равно наибольшему размеру серии продукции. По возможности (в том числе для имитации процесса) следует наполнять средами большее число единиц продукции для обеспечения большей достоверности результатов. Наполнение и герметизация являются, преимущественно, ручными или полуавтоматическими операциями, представляющими риск для стерильности. В связи с этим следует уделить повышенное внимание обучению персонала и аттестации методов асептического производства для конкретных операторов.
Требования к препарату сравнения
19 При существенных изменениях лекарственного средства объем информации о нем (например, по стабильности, сравнительной растворимости, биодоступности) должен быть доступным для демонстрации того, что эти изменения не окажут существенного влияния на исходные параметры качества этого лекарственного средства.
20 Срок годности препарата сравнения, указанный на первоначальной упаковке, не может быть таким же для переупакованного в другую упаковку препарата, поскольку при этом может быть не обеспечен эквивалентный уровень защиты или нарушена совместимость с продуктом. Поэтому спонсор (или лицо, действующее от его имени) обязан определить соответствующий срок годности (с учетом вида препарата, характеристик упаковки и условий хранения). Новый срок годности должен быть обоснован и не может превышать указанный на первоначальной упаковке. Этот срок должен быть совместим со сроком хранения и длительностью клинических исследований.
Операции по кодированию (маскированию)
21 При применении кодирования (маскирования) должна существовать система, гарантирующая маскировку и ее поддерживание. В то же время эта система должна позволять идентифицировать, при необходимости, кодированный ("слепой") препарат, в том числе номера серий до маскирования. Следует предусмотреть возможность быстрого снятия маскировки в экстренных случаях.
Код рандомизации
22 В инструкциях должны быть четко описаны все операции (формирование, распространение, обращение и хранение) с любым кодом рандомизации, используемым для упакованных лекарственных средств для клинических исследований, а также методы снятия кода. Следует ввести соответствующую документацию.
Операции по упаковке
23 При упаковывании лекарственного средства для исследований может оказаться необходимым одновременное обращение с различными продуктами на одной и той же упаковочной линии. Риск перепутывания продуктов должен быть сведен к минимуму за счет применения специальных методов работы и/или специализированного оборудования и обучения персонала.
24 Операции по упаковке и маркировке лекарственных средств для исследований могут быть более сложными и подверженными ошибкам (которые труднее выявлять), чем при производстве для реализации, особенно при кодировании продуктов с похожим внешним видом. В связи с этим требуется принимать особые меры по предотвращению ошибок в маркировке, например, за счет ведения баланса (сопоставления) этикеток, очистки линии, внутрипроизводственного контроля специально подготовленным персоналом.
25 Упаковка должна гарантировать сохранность лекарственного средства в требуемом состоянии при транспортировании и хранении в промежуточных пунктах. Вторичная упаковка должна быть такой, чтобы было сразу заметно любое нарушение ее целостности при транспортировании.
Маркировка
26 В таблице приведены требования, содержащиеся в пунктах 26-30 настоящего приложения. Маркировка должна обеспечивать защиту субъекта исследований, возможность прослеживания и идентификации препарата и испытания и способствовать правильному применению лекарственного средства для исследований*. Маркировка должна содержать следующую информацию, если не обосновано ее отсутствие (например, при наличии централизованной электронной системы рандомизированного кодирования):
________________
* Эти требования установлены ст.15 Директивы 2003/94/ЕС.
a) наименование (имя), адрес и телефон спонсора, контрактной исследовательской организации или исследователя, работающих по контракту (основной контакт для получения информации о препарате, клинических исследованиях и для экстренного декодирования);
b) лекарственную форму, способ введения, количество дозированных единиц, и в случае проведения открытого исследования - наименование/шифр лекарственного средства и его активность/эффективность;
c) номер серии и/или код для идентификации содержимого и операции по упаковке;
d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если это не указано в другом месте;
e) идентификационный номер/лечебный номер субъекта клинического исследования и при необходимости номер посещения;
f) фамилию и инициалы исследователя (если не указано в перечислениях а) или d));
g) инструкции по применению (может быть приведена ссылка на листок-вкладыш, либо другой пояснительный документ, предназначенный для субъекта клинического исследования или лица, которое вводит препарат);
h) надпись "Только для клинических исследований" или аналогичная формулировка;
i) условия хранения;
j) срок использования с указанием месяца и года таким образом, чтобы избежать любой неопределенности (может быть указана дата, до которой необходимо использовать препарат, срок годности или дата повторного контроля);
k) надпись "Хранить в недоступном для детей месте" за исключением случаев, когда препарат предназначен для использования только в условиях стационара.
27 Адрес и номер телефона основного контактного лица для передачи информации относительно препарата, клинического исследования и срочного декодирования могут быть не указаны на этикетке, если субъекту исследования предоставлены листок-вкладыш или карточка, на которой указаны эти данные, а также дана инструкция держать их при себе постоянно.
28 Данные должны быть приведены на официальном языке (языках) страны, где будет применяться лекарственное средство для исследований. Данные, приведенные в пункте 26 данного приложения, должны находиться как на первичной, так и на вторичной упаковке (данные могут быть не указаны на первичной упаковке в случаях, указанных в пунктах 29 и 30 настоящего приложения). Требования к информации, приводимой на первичной и вторичной упаковках, приведены в таблице 13.1. Также на этикетках может быть приведена информация на других языках.
29 Если препарат подготовлен для субъекта исследований или лица, которое вводит препарат, то первичную и вторичную упаковки следует оставлять вместе, и при этом на вторичной упаковке должны содержаться данные, приведенные в пункте 26 настоящего приложения. На этикетке первичной упаковки (или любого укупоренного дозирующего устройства, содержащего первичную упаковку) необходимо указать следующую информацию:
a) наименование (имя) спонсора, исследовательского учреждения или исследователя, работающих по контракту;
b) лекарственную форму, способ введения (можно не указывать для твердых лекарственных форм для орального применения), количество дозированных единиц и в случае проведения открытого исследования, наименование/шифр лекарственного средства, а также активность/эффективность;
c) номер серии и/или код, позволяющие идентифицировать содержимое и операции по упаковке;
d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если все это не указано в другом месте;
e) идентификационный номер/лечебный номер субъекта клинического исследования и, при необходимости, номер его посещения.
30 Если первичной упаковкой является блистер или она имеет малый размер, например ампулы, на которых не могут быть размещены данные, приведенные в пункте 26 настоящего приложения, должна быть предусмотрена вторичная упаковка с этикеткой, содержащей эти данные. Однако на первичной упаковке должны быть указаны:
a) наименование (имя) спонсора, исследовательского учреждения или исследователя, работающих по контракту;
b) способ введения (можно не указывать для твердых лекарственных форм для орального применения) и в случае проведения открытых исследований, наименование/шифр лекарственного средства, а также активность/ эффективность;
c) номер серии и/или код, позволяющие идентифицировать содержимое и операции по упаковке;
d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если все это не указано в другом месте;
e) идентификационный номер/лечебный номер субъекта клинического испытания и при необходимости номер его посещения.
31 Для пояснения указанной выше информации могут быть использованы символы или пиктограммы. Может быть представлена дополнительная информация, предостережение и/или инструкции по обращению с препаратом.
32 В некоторых случаях* при проведении клинических исследований на первичной упаковке дополнительно должны быть приведены следующие данные (так, чтобы они не закрывали первоначальную этикетку):
________________
* Эти случаи указаны в статье 14 Директивы 2001/20/ЕС и относятся к ситуациям, когда:
- нет необходимости в раздельных процессах производства или упаковки;
- при исследовании используются лекарственные средства, зарегистрированные, произведенные или импортированные в соответствии с действующим законодательством;
- в исследованиях принимают участие пациенты с теми заболеваниями, которые соответствуют показаниям к применению, указанным в инструкции по применению, утвержденной при регистрации лекарственного средства.
I - наименование (имя) спонсора, исследовательского учреждения или исследователя, с которым заключен контракт;
II - номер (код) исследования, позволяющий идентифицировать медицинское учреждение, исследователя и субъекта исследований.
33 Если необходимо изменить дату использования лекарственного средства, следует нанести на упаковку дополнительную этикетку. На дополнительной этикетке должна быть указана новая дата, до которой следует использовать препарат, а также повторно указан номер серии. Дополнительную этикетку можно наклеивать поверх старой даты использования, но она не должна закрывать исходный номер серии, что связано с контролем качества. Эту операцию можно осуществлять на производственном участке, который имеет на это право. При необходимости это может осуществляться в исследовательском учреждении фармацевтом, проводящим клинические испытания либо под его наблюдением, а также другим медицинским работником в соответствии с требованиями действующего законодательства. Если это невозможно, операцию может осуществлять лицо, которое прошло соответствующее обучение. Операцию следует осуществлять согласно принципам GMP в соответствии со специальными и стандартными методиками и, при необходимости, по контракту; операцию должно контролировать второе лицо. Дополнительное этикетирование следует тщательным образом документировать как в документах клинических исследований, так и в протоколах серии.
Контроль качества
34 Поскольку процессы могут быть нестандартными или не в полной мере аттестованными, возрастает значение испытаний для обеспечения гарантии того, что каждая серия продукции соответствует спецификации на нее.
35 Контроль качества необходимо осуществлять в соответствии с досье на лекарственное средство и согласно информации, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинических исследований*. Следует проводить проверку эффективности кодирования и результаты ее оформлять документально.
________________
* Эти требования установлены в ст.9(2) Директивы 2001/20/ЕС.
36 Необходимо хранить образцы каждой серии лекарственного средства для исследований, в том числе кодированного препарата, на протяжении не менее пяти лет после завершения или официального прекращения последнего клинического исследования, при проведении которого была использована эта серия*.
________________
* В ЕС эти требования установлены в ст.9 Директивы 2003/94/ЕС.
37 Следует уделить внимание хранению образцов для каждого цикла упаковки/периода испытания до тех пор, пока не будет составлен отчет о клинических исследованиях, чтобы обеспечить возможность подтверждения идентичности препарата при расследовании в случае противоречивых результатов исследований.
Выдача разрешения на выпуск серий
38 Не допускается выдача разрешения на выпуск лекарственных средств для исследований (см. пункт 43 настоящего приложения) до тех пор, пока уполномоченное лицо не удостоверит выполнение установленных требований и требований данного приложения* (см. пункт 39 настоящего приложения). Уполномоченное лицо должно учитывать факторы, приведенные в пункте 40 настоящего приложения.
________________
* В ЕС требования к выдаче разрешения на выпуск лекарственных средств, предназначенных для исследований, уполномоченным лицом изложены в статье 13.3 Директивы 2001/20/ЕС.
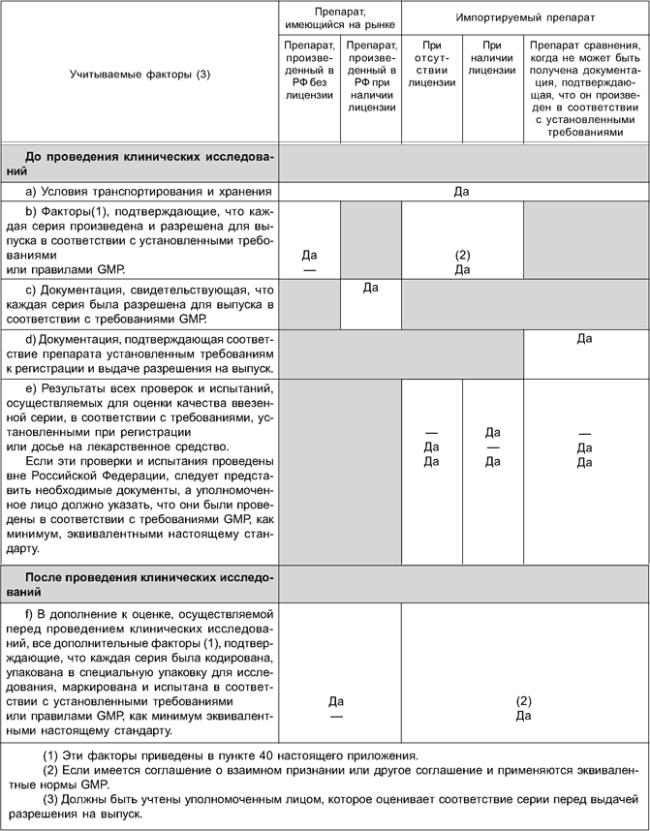
39 На выполнение уполномоченным лицом своих обязанностей в отношении лекарственных средств для исследований влияют разные факторы.
В большинстве случаев необходимо учитывать следующее (таблица 13.2):
a) препарат произведен в Российской Федерации, но не зарегистрирован в Российской Федерации: необходимо указать, что лекарственные средства для исследований произведены и проверены в соответствии с требованиями GMP, изложенными в настоящем стандарте, досье на лекарственное средство и информацией, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинических исследований*;
________________
* См. статью 13.3, перечисление а) Директивы 2001/20/ЕС.
b) препарат находится на рынке Российской Федерации, поставляется производителем или дистрибьютором, который имеет на то право, и зарегистрирован в Российской Федерации независимо от того, где производится препарат. Его обязанности указаны выше, но объем представленных данных может быть ограничен подтверждением того, что препараты соответствуют уведомлению/запросу на разрешение проведения клинических исследований и любой последующей обработки с целью кодирования, специальной упаковки или маркировки для этого исследования. Досье на лекарственное средство также может быть ограниченным по объему (см. пункт 9 данного приложения);
c) препарат ввезен из другой страны: необходимо указать, что он произведен и проверен в соответствии с требованиями GMP (как минимум, эквивалентными изложенным в настоящем стандарте), досье на лекарственное средство и информацией, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинического исследования*. Если лекарственные средства для исследований ввезены из другой страны и являются объектом соглашения, принятого между Российской Федерацией и этой страной, например таким, как соглашение о взаимном признании, любое подобное соглашение предусматривает применение эквивалентных стандартов GMP в отношении этого препарата. При отсутствии соглашения о взаимном признании уполномоченное лицо на основе информации о системе качества производителя должно установить, что применяются эквивалентные стандарты GMP. Эту информацию, как правило, получают путем участия в аудите систем качества производителей. И в первом, и во втором случае уполномоченное лицо может выполнить оценку соответствия на основании документации, предоставленной производителем из другой страны (см. пункт 40 данного приложения);
________________
* См. статью 13.3, перечисление b) Директивы 2001/20/ЕС.
d) При ввозе препаратов сравнения, когда невозможно получить гарантию того, что каждая серия продукции была произведена в соответствии с эквивалентными стандартами GMP, уполномоченное лицо должно указать, что каждая произведенная серия прошла все необходимые виды контроля и испытаний, необходимые для подтверждения ее качества в соответствии с информацией, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинических исследований*.
________________
* См. статью 13.3, перечисление с) Директивы 2001/20/ЕС.
40 При оценке каждой серии продукции перед выдачей разрешения на выпуск следует рассматривать:
- протоколы серии, в том числе протоколы контроля качества, протоколы внутрипроизводственного контроля и документацию, свидетельствующую о соответствии досье на лекарственное средство заказу и коду рандомизации. В эти протоколы должны быть внесены все отклонения или внесенные в плановом порядке изменения, а также любые дополнительные проверки или испытания. Протоколы должны быть полными и согласованными с персоналом, уполномоченным на это в соответствии с системой качества;
- условия производства;
- данные об аттестации (испытаниях) технических средств, процессов и методов;
- проверку окончательной упаковки;
- результаты любых анализов или испытаний, проведенных после импортирования, если необходимо;
- отчеты о стабильности;
- данные о поставщике и проверке условий хранения и транспортирования;
- отчеты об аудитах системы качества производителя;
- документы, подтверждающие право производителя на производство лекарственных средств для исследований, или соответствующие документы на экспорт, выданные компетентными органами страны-экспортера;
- при необходимости, нормативные требования в регистрационной документации (лицензии) по применению стандартов GMP и любые официальные подтверждения выполнения требований GMP;
- все другие факторы, которые уполномоченное лицо считает значимыми для качества серии.
Значение вышеприведенных факторов зависит от страны, в которой производят препарат, предприятия-производителя, статуса препарата на рынке (является ли он зарегистрированным, зарегистрирован ли он в РФ или в других странах), а также от фазы разработки.
Спонсор должен убедиться в том, что все факторы, принятые во внимание уполномоченным лицом, выполняющим оценку серии, соответствуют информации, предоставленной компетентному органу при запросе разрешения на проведение клинических исследований (см. также пункт 44 данного приложения).
41 Если лекарственные средства для исследований производят и упаковывают на разных участках, за которые несут ответственность разные уполномоченные лица, необходимо выполнять требования приложения 16 к настоящему стандарту.
42 Если согласно действующему законодательству упаковка и маркировка осуществляются в исследовательском учреждении фармацевтом, проводившим клинические исследования, под его наблюдением, либо другим медицинским работником, то контроль этой деятельности не входит в обязанности уполномоченного лица. Однако спонсор несет ответственность за документальное оформление работы и выполнение ее в соответствии с требованиями GMP. По этому вопросу он должен получить информацию от уполномоченного лица.
Транспортирование
43 Транспортирование препаратов для исследований следует осуществлять в соответствии с инструкциями, предоставленными в распоряжение спонсором или лицом, действующим от его имени.
44 Лекарственные средства для исследований следует хранить под контролем спонсора до завершения двух этапов процедуры выдачи разрешения на выпуск: оценки соответствия уполномоченным лицом и выдачи разрешения на выпуск после соблюдения требований действующего законодательства*.
________________
* В ЕС этот порядок регулируется Директивой 2001/20/ЕС (статья 9).
45 До поставки лекарственных средств для исследований к месту проведения исследований должны быть заключены соглашения по декодированию уполномоченным на то персоналом.
46 Следует хранить подробный перечень отгруженной продукции, составленный производителем или импортером. Особое внимание следует уделять точности указания наименования и адреса получателя.
47 Передачу лекарственных средств для исследований из одного места проведения исследований на другое следует проводить только в исключительных случаях. Порядок передачи должен быть установлен инструкцией. Следует проверить историю лекарственного средства за тот период, когда он находился вне контроля производителя, например, с помощью отчетов о клинических исследованиях или документов об условиях хранения на предыдущем месте проведения исследований. Такая проверка должна учитываться при оценке стабильности продукции, предназначенной для передачи. К участию в проверке необходимо привлекать уполномоченное лицо. При необходимости, препарат следует вернуть производителю или другому имеющему на то право производителю для повторной маркировки и для его оценки уполномоченным лицом. Следует хранить протоколы и обеспечивать полное отслеживание подобных передач.
Рекламации
48 Результаты любого расследования, связанного с качеством препарата, должны быть рассмотрены производителем или импортером и спонсором. В этом должны участвовать уполномоченное лицо и лица, ответственные за проведение клинических исследований, чтобы оценить возможное влияние на испытания, разработку препарата и субъектов исследований.
Отзывы и возвраты
Отзывы
49 Порядок возврата лекарственных средств для исследований и его документального оформления должен быть согласован между спонсором и производителем или импортером. Исследователь и другие лица должны понимать свои обязанности при выполнении возврата.
50 Спонсор должен убедиться в наличии у поставщика препарата сравнения или других лекарств, используемых в клинических исследованиях, а также системы для извещения спонсора о необходимости отзыва любой серии препарата.
Возвраты
51 Лекарственные средства для исследований следует возвращать в соответствии с требованиями спонсора и инструкцией.
52 Возвращенные лекарственные средства для исследований должны быть четко обозначены. Их следует хранить в специально отведенной контролируемой зоне. Следует сохранять документацию на возвращенные лекарственные средства.
Уничтожение
53 Спонсор несет ответственность за уничтожение неиспользованных и/или возвращенных лекарственных средств для исследований. Не допускается уничтожение лекарственных средств для клинических исследований без получения разрешения от спонсора.
54 Для каждого места проведения исследований и каждого периода исследований спонсор или лицо, действующее от его имени, должен составлять баланс и проверять количество препарата, которое поставлено, использовано и возвращено. Уничтожение неиспользованных лекарственных средств для исследований для данного места проведения исследований или данного периода исследований следует осуществлять только после проведения расследования, предоставления объяснений любых несоответствий и составления баланса. Документальное оформление операций по уничтожению препарата необходимо вести таким образом, чтобы можно было представить отчет о всех операциях. Протоколы следует хранить у спонсора.
55 В случае уничтожения препаратов, спонсору должен быть представлен акт с указанием даты или другой документ об уничтожении. В этих документах следует четко указать номера серий и/или номера пациентов (или обеспечить возможность их отслеживания) и количество уничтоженных препаратов.
Таблица 13.1 - Общая информация о маркировке (пункты 26-30)

Таблица 13.2 - Информация о выдаче разрешения на выпуск серии лекарственного средства для исследований