
ПРИЛОЖЕНИЕ 2
Обязательное
ИСО 6353-1-82
Реактивы для химического анализа. Часть 1. Общие методы испытаний (ОМ)
5. Общие методы анализа (ОМ)
5.3. Определение массовой доли сульфатов (ОМ 3)
Готовят затравочный раствор, смешивая 0,25 см раствора сульфата калия с массовой долей 0,02% в растворе этанола с объемной долей 30% и 1 см раствора 2-водного хлорида бария с массовой долей 25%. Ровно через 1 мин к этой смеси добавляют указанный объем анализируемого раствора (Р 16.3.4), предварительно подкисленный 0,5 см раствора соляной кислоты с массовой долей 20%.
раствора сульфата калия с массовой долей 0,02% в растворе этанола с объемной долей 30% и 1 см раствора 2-водного хлорида бария с массовой долей 25%. Ровно через 1 мин к этой смеси добавляют указанный объем анализируемого раствора (Р 16.3.4), предварительно подкисленный 0,5 см раствора соляной кислоты с массовой долей 20%.
Смесь отстаивают в течение 5 мин и сравнивают ее помутнение с помутнением смеси, полученной при аналогичной обработке соответствующего контрольного раствора.
5.4. Определение массовой доли фосфатов (ОМ 4)
К указанному объему анализируемого раствора (Р 16.3.3) добавляют 5 см раствора молибдата аммония с массовой долей 10%. рН раствора доводят до 1,8 и нагревают до кипения. Охлаждают, добавляют 12,5 см раствора соляной кислоты с массовой долей 15% и экстрагируют 20 см диэтилового эфира. Органический слой промывают раствором соляной кислоты с массовой долей 5% и восстанавливают молибдено-фосфатный комплекс 0,2 см раствора 2-водного хлорида олова (II) с массовой долей 2% в соляной кислоте. Сравнивают интенсивность синей окраски полученного органического слоя с интенсивностью окраски органического слоя, полученного при аналогичной обработке соответствующего контрольного раствора.
5.6. Определение массовой доли общего азота (ОМ 6)
К указанному объему анализируемого раствора (Р 16.3.5), разбавленному при необходимости до 140 см, в приборе Кьельдаля, состоящем из колбы Кьельдаля и перегонного устройства, добавляют 5 см раствора гидроксида натрия с массовой долей 32% и 1,0 г сплава Деварда или алюминиевой проволоки. Выдерживают в течение 1 ч. Отгоняют 75 см реакционной смеси в мерный цилиндр, содержащий 5,0 см раствора серной кислоты с массовой долей 0,5%. Добавляют 3 см раствора гидроксида натрия с массовой долей 32%, 2 см реактива Несслера и разбавляют до 100 см.
Сравнивают интенсивность желтой окраски полученного раствора с интенсивностью окраски раствора, полученного при аналогичной обработке соответствующего контрольного раствора.
5.29. Атомно-абсорбционная спектроскопия (ОМ 29)
5.29.1. Общие указания
Испытуемый образец или его раствор распыляют в высокотемпературное пламя, создаваемое смесью горючего газа и газа-окислителя, обеспечивающее испарение испытуемого образца и диссоциацию его молекул на атомы. Может быть использован прибор с беспламенным нагревом. Источник излучения, представляющий собой электронную лампу с полым катодом или безэлектродную разрядную трубку, активируемую микроволновым излучением, продуцирует излучение с длиной волны, соответствующей энергии возбуждения атомов испытуемого вещества. Атомы определяемого элемента поглощают определенную долю этого излучения, пропорциональную их количеству в основном (невозбужденном) состоянии, и это поглощение регистрируется атомно-абсорбционным спектрометром.
5.29.2. Методика анализа
Сущность метода, многообразие существующих приборов, обилие параметров, связанных с испытуемым образцом и с прибором, и множественность влияющих факторов не позволяют дать подробных инструкций.
Выбор методики определяется требуемой степенью точности.
Следует принимать во внимание возможность возникновения помех от пламенных и беспламенных источников нагрева. Если прибор укомплектован пламенным источником нагрева, определение проводят, используя водные растворы испытуемых веществ, слегка подкисленные азотной или соляной кислотой.
В целях учета эффектов раствора рекомендуется пользоваться методом добавок. Этот метод состоит в том, что определение осуществляют для серии (размер которой зависит от требуемой точности, но не меньше двух) растворов испытуемого образца, к которым добавлены известные количества определяемого вещества.
Длины волн, соответствующие резонансным линиям, и другая специальная информация приводятся в описаниях, относящихся к определенному реактиву.
5.30. Пламенная фотометрия (ОМ 30)
5.30.1. Общие указания
Метод основан на измерении интенсивности светового излучения, испускаемого некоторыми атомами при переходе из возбужденного состояния в состояние с более низкой энергией. Атомы переходят в возбужденно состояние в пламени, создаваемом смесью горячего газа и газа-окислителя. Интенсивность испускаемого атомами излучения измеряют с помощью фотометрической системы либо с монохроматором, либо с фильтрами.
Примечание. Могут быть использованы отличные от указанных в описаниях смеси газов для пламени, при этом может возникнуть необходимость изменить рекомендованные в этих же описаниях концентрации растворов.
5.30.2. Методика анализа
Методика анализа сходна с методикой атомно-абсорбционной спектроскопии. Условия для каждого конкретного анализа можно найти в описаниях, касающихся анализируемого реактива.
5.31.1. Определение рН (ОМ 31.1)
5.31.1.1. Общие положения
Рассмотрим гальванический элемент: электрод сравнения - насыщенный раствор  -раствор
-раствор 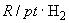 . Для буферных растворов
. Для буферных растворов  и
и  с известными значениями рН, соответственно
с известными значениями рН, соответственно  и
и 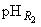 , измеренные значения разности потенциалов составляют соответственно
, измеренные значения разности потенциалов составляют соответственно  и
и  .
.
Если раствор  в рассматриваемом гальваническом элементе заменить исследуемым раствором с неизвестным рН, то по разности измеренных значений потенциалов можно рассчитать рН исследуемого раствора.
в рассматриваемом гальваническом элементе заменить исследуемым раствором с неизвестным рН, то по разности измеренных значений потенциалов можно рассчитать рН исследуемого раствора.
Если все измерения проведены при одной и той же температуре и при неизменной концентрации раствора хлорида калия, рН исследуемого раствора может быть рассчитан по следующим формулам:
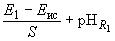 ;
;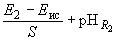 ;
;
где  - электродвижущая сила гальванического элемента с исследуемым раствором;
- электродвижущая сила гальванического элемента с исследуемым раствором; - угловой коэффициент:
- угловой коэффициент:
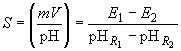 .
.
5.31.1.2. Аппаратура
рН-метр со стеклянным электродом, соединенным с миливольтметром с большим сопротивлением и со шкалой, откалиброванной в единицах рН. Такой прибор, регистрируя разность потенциалов между рН-чувствительным электродом (стеклянным, сурьмяным) и электродом сравнения, соединенным электролитическим мостиком (например, насыщенный раствор ), дает возможность непосредственно считывать со шкалы значения рН.
5.31.1.3. Калибровка
рН-метр калибруют, используя буферные растворы с известной активностью ионов водорода, например:
а) оксалатный буферный раствор;
б) тартратный буферный раствор;
в) фталатный буферный раствор;
г) фосфатный буферный раствор;
д) боратный буферный раствор;
е) буферный раствор гидроксида кальция.
В табл.3 приведены значения рН перечисленных буферных растворов в интервале температур 15-35 °С.
Таблица 3
Температура, °С |
рН буферного раствора |
|||||
а |
б |
в |
г |
д |
е |
|
15 |
1,67 |
- |
4,00 |
6,90 |
9,27 |
12,81 |
20 |
1,68 |
- |
4,00 |
6,88 |
9,22 |
12,63 |
25 |
1,68 |
3,56 |
4,01 |
6,86 |
9,18 |
12,45 |
30 |
1,69 |
3,55 |
4,01 |
6,85 |
9,14 |
12,30 |
35 |
1,69 |
3,55 |
4,02 |
6,84 |
9,10 |
12,14 |
5.31.1.4. Методика анализа
Готовят анализируемый раствор (кроме тех случаев, когда анализируют непосредственно сам реактив) заданной концентрации, применяя воду, свободную от диоксида углерода.
Одновременно готовят два буферных раствора, среднее значение рН которых примерно равно предполагаемому значению рН анализируемого раствора. Температуру всех трех растворов, а также ячейки прибора устанавливают равной (25±1) °С.
Прибор калибруют с помощью двух буферных растворов, промывая измерительный электрод перед измерением буферным раствором. После промывания электрода водой и анализируемым раствором измеряют рН анализируемого раствора.
Для получения точных результатов необходимо повторять измерения с различными порциями анализируемо раствора без промывания электрода между последовательными измерениями до тех пор, пока рН не будет охраняться постоянным не менее 1 мин.
5.35. Определение металлов путем экстракции с последующей атомно-абсорбционной спектроскопией (ОМ 35)
Готовят 150 см анализируемого раствора, добавляя соответствующий объем уксусной кислоты или раствора гидроксида натрия с массовой долей 20% для доведения рН раствора до 5. Полученный раствор разделяют на три равные порции, которые помещают в три делительные воронки. В две из них добавляют контрольные растворы определяемых металлов, в первую - в количестве, эквивалентном предполагаемой предельной массе металла в анализируемом растворе, а во вторую - вдвое большем количестве. Содержимое каждой из трех делительных воронок обрабатывают следующим образом: добавляют 1 см раствора пирролидин-карбодитиоата аммония с массовой долей 1%, перемешивают, добавляют 10 см 4-метилпентан-2-она и встряхивают 30 с. После разделения фаз водный слой отбрасывают. Органический слой переносят в мерную колбу на 10 см и разбавляют до метки этанолом с объемной долей 95%. Полученные экстракты анализируют в соответствии с ОМ 29.
ПРИЛОЖЕНИЯ 1, 2. (Введены дополнительно, Изм. N 3).
Электронный текст документа
подготовлен Free Of Charge Document и сверен по:
официальное издание
М.: ИПК Издательство стандартов, 1998