
ОСНОВНЫЕ ТРЕБОВАНИЯ К АКТИВНЫМ ФАРМАЦЕВТИЧЕСКИМ СУБСТАНЦИЯМ (АФС), ИСПОЛЬЗУЕМЫМ В КАЧЕСТВЕ ИСХОДНЫХ МАТЕРИАЛОВ
1 Введение
1.1 Цель
1.2 Область применения
2 Обеспечение качества
2.1 Общие положения
2.2 Функции и ответственность подразделений по обеспечению и контролю качества
2.3 Функции и ответственность производственных подразделений
2.4 Внутренние аудиты (самоинспекции)
2.5 Анализ качества продукции
3 Персонал
3.1 Квалификация персонала
3.2 Гигиена персонала
3.3 Консультанты
4 Здания, помещения и инженерные системы
4.1 Проектирование и строительство
4.2 Инженерные системы
4.3 Подготовка воды
4.4 Разделение зон
4.5 Освещение
4.6 Стоки и отходы
4.7 Уборка, дезинфекция и техническое обслуживание
5 Технологическое оборудование
5.1 Требования к конструкции и монтажу
5.2 Техническое обслуживание и очистка оборудования
5.3 Калибровка (поверка)
5.4 Системы с компьютерным управлением и контролем
6 Документация и протоколы
6.1 Система документации и спецификации
6.2 Протокол очистки и использования оборудования
6.3 Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы для АФС
6.4 Промышленные регламенты
6.5 Протоколы на серию продукции (протоколы производства и контроля качества серии продукции)
6.6 Протоколы лабораторного контроля
6.7 Рассмотрение протоколов на серию продукции
7 Работа с материалами
7.1 Общий контроль
7.2 Приемка и карантин
7.3 Отбор проб и проведение испытаний поступивших материалов
7.4 Хранение
7.5 Повторный контроль
8 Технологический процесс и внутрипроизводственный контроль
8.1 Технологические операции
8.2 Ограничения на время выполнения операций
8.3 Внутрипроизводственный отбор проб и контроль
8.4 Смешанные серии промежуточных продуктов или АФС
8.5 Контроль загрязнений
9 Упаковка и маркировка АФС и промежуточных продуктов
9.1 Общие положения
9.2 Упаковочные материалы
9.3 Выпуск и контроль печатных материалов
9.4 Операции по упаковке и маркировке
10 Хранение и реализация
10.1 Хранение на складе
10.2 Реализация
11 Лабораторный контроль
11.1 Общий контроль
11.2 Проведение испытаний промежуточных продуктов и АФС
11.3 Аттестация аналитических методик
11.4 Аналитические паспорта
11.5 Контроль стабильности АФС
11.6 Срок годности и дата повторного контроля
11.7 Архивные образцы
12 Аттестация (испытания)
12.1 Политика проведения аттестации (испытаний)
12.2 Документация по аттестации (испытаниям)
12.3 Этапы аттестации (испытаний)
12.4 Аттестация (испытания) процесса
12.5 Программа аттестации (испытаний) процесса
12.6 Периодическая оценка аттестованных (испытанных) систем и процессов
12.7 Аттестация методов очистки
12.8 Аттестация аналитических методов
13 Контроль изменений
14 Отклонение и переработка материалов
14.1 Отклонение
14.2 Повторная обработка
14.3 Переработка
14.4 Регенерация материалов и растворителей
14.5 Возврат
15 Рекламации и отзывы
16 Работа по контракту (в т.ч. проведение анализов)
17 Реализация, хранение, переупаковка и перемаркировка
17.1 Область применения
17.2 Прослеживаемость реализуемых промежуточных продуктов или АФС
17.3 Обеспечение качества
17.4 Переупаковка, перемаркировка и обращение с промежуточными продуктами или АФС
17.5 Стабильность
17.6 Передача информации
17.7 Работа с рекламациями и отзывами
17.8 Работа с возвратами
18 АФС, производимые путем культивирования клеток (ферментации)
18.1 Общие положения
18.2 Поддерживание банков клеток и ведение протоколов
18.3 Культивирование клеток (ферментация)
18.4 Сбор, выделение и очистка
18.5 Удаление вирусов (стадии инактивации)
19 АФС, предназначенные для проведения клинических исследований
19.1 Общие положения
19.2 Качество
19.3 Оборудование
19.4 Контроль исходных материалов
19.5 Производство
19.6 Аттестация (испытания)
19.7 Изменения
19.8 Лабораторный контроль
19.9 Документация
20 Термины и определения
1.1 Цель
Данная часть стандарта является руководством по применению правил GMP к производству активных фармацевтических субстанций (АФС) с целью обеспечения гарантии их качества и соответствия требованиям к чистоте.
Термин "производство" включает в себя все виды операций с АФС: приемку исходного сырья, производство, упаковку, переупаковку, маркировку, перемаркировку, контроль качества, выпуск продукции, хранение и реализацию, а также соответствующие меры контроля. Понятия "должен", "следует", применяемые в настоящем руководстве, указывают на рекомендации, выполнение которых предполагается, за исключением случаев, когда их выполнить невозможно или они могут быть заменены альтернативными действиями, по крайней мере, с эквивалентным уровнем обеспечения качества продукции.
Стандарт не затрагивает вопросов безопасности персонала, занятого в производстве, и требований по защите окружающей среды. Производитель несет ответственность за безопасность персонала и окружающей среды в соответствии с законодательством.
Стандарт не устанавливает требований, предъявляемых при регистрации (подаче заявки на регистрацию) АФС, и не заменяет требований Фармакопеи, не затрагивает полномочий соответствующих органов по установлению специфических требований к АФС для выдачи разрешения на реализацию/производство или применение лекарственных средств. Следует выполнять все требования, установленные при государственной регистрации АФС.
1.2 Область применения
Стандарт устанавливает требования к производству АФС, используемых в лекарственных средствах для человека и животных (медицинских препаратах). К производству стерильных АФС оно применимо только до стадии стерилизации. Стандарт не распространяется на процессы стерилизации и производство стерильных АФС в асептических условиях. Эти процессы следует проводить в соответствии с требованиями настоящего стандарта (в т.ч. приведенными в приложении 1) и других нормативных документов.
В случае производства средств против эктопаразитов могут использоваться другие нормативные документы.
Стандарт не распространяется на производство цельных клеток, цельной крови и плазмы, производных крови и плазмы (фракционирование плазмы), но распространяется на производство АФС, получаемых с использованием крови или плазмы в качестве исходного сырья. На производство клеточных субстратов (млекопитающих, растений, насекомых или бактерий, тканей или органов животных, в т.ч. трансгенных животных) и ранние стадии производства могут распространяться требования настоящего стандарта, выходящие за рамки данной части стандарта. Стандарт не распространяется на производство нерасфасованных лекарственных средств, но распространяется на активные фармацевтические субстанции, относящиеся к приложениям 2-7 настоящего стандарта.
Раздел 19 стандарта содержит требования, распространяющиеся только на производство АФС, используемых для получения лекарственных средств, предназначенных для клинических исследований (исследовательских лекарственных средств).
Исходный материал для производства АФС - сырье, промежуточные продукты или другие АФС, используемые в производстве АФС и включаемые в АФС в качестве существенного структурного элемента. Исходный материал для производства АФС может быть продуктом (материалом), получаемым от одного или более поставщиков, или материалом, производимым на самом предприятии. Как правило, для производства АФС используются исходные материалы с конкретными химическими свойствами и структурой.
Производитель АФС должен определить и документально оформить стадию, с которой должно начинаться производство АФС из исходного сырья. Для процессов химического синтеза эта стадия определяется как стадия ввода исходных материалов в технологический процесс производства АФС. Для других процессов (ферментации, экстракции, очистки и пр.) данную стадию определяют с учетом конкретных особенностей производства. В таблице 1 приведены стадии производства различных АФС с выделением стадий, на которых исходный материал, как правило, вводится в технологический процесс.
Начиная с этой стадии, на данные промежуточные продукты и/или стадии производства АФС действуют правила данной части стандарта. Они включают в себя требования к аттестации (испытаниям) критических стадий технологического процесса, оказывающих влияние на качество АФС. В то же время выбор стадий технологического процесса для проведения аттестации (испытаний) не обязательно означает, что эти стадии являются критическими для качества АФС.
Требования настоящей части распространяются, как правило, на стадии, выделенные в таблице 1 серым фоном. Это не означает, что в процессе производства должны выполняться все стадии, указанные в данной таблице. Строгость следования требованиям GMP должна возрастать от ранних стадий производства АФС к завершающим стадиям технологического процесса, очистки и упаковки. Физическую обработку АФС, такую как грануляция, покрытие оболочкой или физическое изменение размера частиц (например, грубый и тонкий помол), следует проводить, по крайней мере, в соответствии с требованиями настоящего стандарта.
Таблица 1 - Применение стандарта для производства АФС
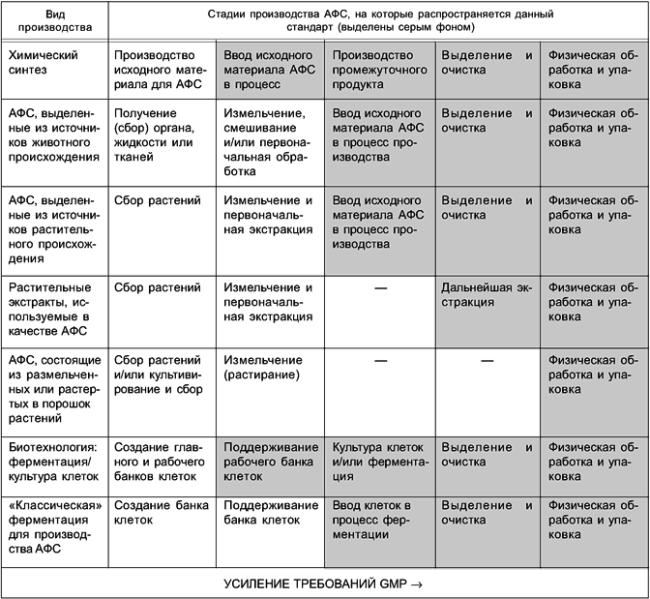
Настоящий стандарт не распространяется на технологические стадии, предшествующие вводу исходных материалов для производства АФС в технологический процесс.
Термин "активная фармацевтическая субстанция" может использоваться наряду с термином "активная субстанция". Термины и определения, приведенные в разделе 20 части II, относятся только к этой части. Ряд терминов определен в части I и должен использоваться в том же смысле.
2.1 Общие положения
2.10 Ответственность за качество выпускаемого продукта несет весь персонал предприятия-производителя.
2.11 На каждом предприятии следует разработать, документально оформить и внедрить систему обеспечения качества, которая предусматривает активное участие в ней руководителей предприятия и всего персонала, занятого в производстве.
2.12 Система обеспечения качества охватывает организационную структуру, инструкции, методики, процессы и ресурсы, а также действия, необходимые для обеспечения гарантии соответствия АФС всем требованиям к качеству и чистоте. Деятельность, связанная с обеспечением качества, должна быть четко определена и документально оформлена.
2.13 Подразделения (отделы, службы), ответственные за обеспечение и контроль качества, должны быть независимыми от производства. Эти функции могут выполнять отдельные подразделения по обеспечению и контролю качества либо быть возложены на одно лицо (группу лиц) в зависимости от объемов и структуры предприятия.
2.14 Следует определить круг лиц, ответственных за выпуск промежуточных продуктов и АФС.
2.15 Все действия по контролю качества следует оформлять документально непосредственно при их выполнении.
2.16 Любые отклонения от принятых инструкций должны быть обоснованы и оформлены документально. Критические отклонения следует расследовать с необходимым документальным оформлением.
2.17 Не допускается выпускать или использовать материалы до получения положительного заключения отдела(ов) качества, если не установлен специальный порядок, допускающий соответствующие отклонения (например, выпуск материалов, находящихся в карантине, приведенных в 10.20 настоящего руководства, или использование сырья или промежуточных продуктов, оценка качества которых еще не завершена).
2.18 Следует разработать порядок своевременного извещения руководства предприятия о результатах инспекций, проводимых органами контроля и надзора, случаях серьезного нарушения требований настоящего стандарта, обнаружения несоответствия продукта и принятых мерах (претензиях к качеству продукции, отзывах продукции, заключениях инспекции и т.д.).
2.2 Функции и ответственность подразделений по обеспечению и контролю качества
2.20 Деятельность подразделений по обеспечению и контролю качества распространяется на все аспекты качества.
2.21 Подразделения по обеспечению и контролю качества должны рассматривать и согласовывать (утверждать) все документы, имеющие отношение к качеству продукции.
2.22 Функции и ответственность службы качества, независимой от производства, не могут быть переданы другим подразделениям или лицам. Функции и ответственность этой службы должны быть оформлены документально и включать в себя, по крайней мере, следующее:
1 Выпуск или отзыв любых АФС. Выпуск или отзыв промежуточных продуктов, используемых вне зоны контроля предприятия-производителя.
2 Разработку и ввод в действие системы выпуска или отзыва сырья, промежуточных продуктов, упаковочных и печатных материалов (материалов для маркировки).
3 Проверки серии готовой продукции и протоколов лабораторных анализов на критических стадиях технологического процесса до выпуска АФС в реализацию.
4 Расследование причин критических отклонений параметров от установленных значений с принятием необходимых мер и т.д.
5 Согласование всех спецификаций и технологических инструкций по производству продукции.
6 Согласование всех инструкций и методик по качеству промежуточных продуктов или АФС.
7 Контроль проведения внутренних проверок (самоинспекций).
8 Согласование производителей промежуточных продуктов и АФС, работающих по контракту.
9 Согласование изменений, которые могут оказать влияние на промежуточные продукты и АФС.
10 Рассмотрение и согласование протоколов и отчетов о проведении аттестации (испытаний).
11 Контроль расследования претензий, связанных с качеством продукции, и принятия необходимых мер.
12 Контроль своевременного выполнения технического обслуживания и калибровки (поверки) критического оборудования в соответствии с установленным порядком.
13 Контроль проведения испытаний и оформления их результатов.
14 Контроль (при необходимости) данных о стабильности АФС и/или промежуточных продуктов для обоснования даты их повторного контроля, срока годности и условий их хранения.
15 Составление отчетов о качестве продукции (подраздел 2.5 настоящего руководства).
2.3 Функции и ответственность производственных подразделений
Функции и ответственность производственных подразделений должны быть оформлены документально и включать в себя, по крайней мере, следующее:
1 Разработку, пересмотр и согласование (утверждение) инструкций по производству промежуточных продуктов и/или АФС, а также обеспечение инструкциями всех причастных подразделений и исполнителей в соответствии с утвержденной инструкцией.
2 Производство АФС и, при необходимости, промежуточных продуктов в соответствии с утвержденными инструкциями.
3 Рассмотрение всех протоколов производства серии продукции и подтверждение их полноты и правильности оформления (подписания) в установленном порядке.
4 Контроль проведения анализа всех отклонений в процессе производства и расследований критических отклонений с соответствующим документальным оформлением.
5 Контроль чистоты производственных помещений и, при необходимости, их дезинфекции.
6 Контроль выполнения необходимых калибровок (поверок) и хранения протоколов калибровки.
7 Контроль содержания помещений и оборудования в надлежащем состоянии и хранения соответствующих протоколов.
8 Контроль ведения и согласования (утверждения) протоколов и отчетов аттестации (испытаний) оборудования и процессов.
9 Оценка предлагаемых изменений в продукции, технологическом процессе или оборудовании.
10 Контроль проведения аттестации новых и, при необходимости, модернизированных помещений и оборудования.
2.4 Внутренние аудиты (самоинспекции)
2.40 Для подтверждения соответствия производства АФС требованиям настоящего стандарта следует регулярно проводить внутренние аудиты согласно утвержденному графику.
2.41 Результаты аудита и последующие корректирующие действия следует оформлять документально и представлять руководству предприятия. Действия по результатам аудита следует выполнять своевременно и надлежащим образом.
2.5 Анализ качества продукции
2.50 Для подтверждения постоянного соответствия технологического процесса установленным требованиям следует регулярно проводить анализ качества АФС. Такой анализ следует проводить, как правило, ежегодно с последующим документальным оформлением. Отчет о проведении анализа качества продукции должен включать в себя, по крайней мере, следующее:
- рассмотрение результатов внутрипроизводственного контроля и испытаний АФС по критическим параметрам;
- анализ данных о всех сериях продукции, качество которых не соответствовало установленным требованиям;
- анализ всех существенных отклонений в технологическом процессе и результаты расследования их причин;
- рассмотрение всех изменений, внесенных в технологический процесс или аналитические методы;
- рассмотрение результатов непрерывного контроля стабильности;
- анализ всех претензий, возвратов и отзывов, связанных с качеством продукции;
- анализ эффективности всех корректирующих действий, принятых по результатам аудита.
2.51 Следует оценить результаты анализа и принять решение о необходимости принятия соответствующих мер или повторной аттестации (испытаний). Обоснование необходимости этих мер должно быть оформлено документально. Указанные меры должны быть выполнены своевременно и надлежащим образом.
3.1 Квалификация персонала
3.10 Производство должно быть укомплектовано необходимым числом сотрудников, имеющих соответствующее образование, прошедших обучение и/или имеющих достаточный опыт работы в производстве АФС и промежуточных продуктов.
3.11 Ответственность и обязанности всех сотрудников, работающих в производстве АФС и промежуточных продуктов, должны быть документально оформлены.
3.12 Следует проводить регулярное обучение персонала с привлечением специалистов необходимой квалификации. Это обучение должно охватывать, как минимум, вопросы, связанные с выполнением конкретных операций и требований настоящего стандарта в части обязанностей данного сотрудника. Следует вести и сохранять протоколы обучения сотрудников, а также проводить периодическую оценку их квалификации.
3.2 Гигиена персонала
3.20 Персонал должен соблюдать правила гигиены.
3.21 Персонал должен носить чистую одежду, соответствующую его производственной деятельности. Следует определить порядок замены этой одежды. Для защиты АФС и промежуточных материалов от загрязнения, при необходимости, следует носить дополнительные элементы одежды, покрывающие голову, лицо и руки.
3.22 Следует избегать прямого контакта персонала с промежуточными продуктами и АФС.
3.23 Курение, прием пищи и напитков, жевание резинки и хранение продуктов питания допускаются только в специально выделенных местах, отделенных от производственных помещений.
3.24 Сотрудники с инфекционными заболеваниями или имеющие открытые повреждения на незакрытых участках тела не допускаются к работе, если это может отразиться на качестве АФС. Любой сотрудник с заболеванием или наличием открытой раны, выявленными при медицинском осмотре или наблюдении, должен быть отстранен от производственных операций, при выполнении которых состояние его здоровья может неблагоприятно повлиять на качество АФС, до выздоровления или получения медицинского заключения о том, что нахождение этого сотрудника на рабочем месте не представляет риска для безопасности или качества АФС.
3.3 Консультанты
3.30 Консультанты по вопросам производства и контроля качества промежуточных материалов или АФС должны иметь соответствующее образование, подготовку и опыт работы или сочетание вышеперечисленных требований.
3.31 Следует организовать ведение и хранение протоколов о проведении консультаций с указанием имен, адресов, специальностей и видов услуг, предоставляемых этими консультантами.
4.1 Проектирование и строительство
4.10 При размещении, проектировании и строительстве зданий и помещений, предназначенных для производства промежуточных продуктов и АФС, следует предусматривать удобство их эксплуатации и обслуживания в соответствии с видом и стадией производства. При проектировании помещений следует сводить к минимуму возможность загрязнения. Если для промежуточных продуктов и АФС установлены требования к микробной чистоте, то при проектировании помещений следует предусматривать защиту от микробных загрязнений.
4.11 Здания и помещения должны иметь достаточные размеры для размещения оборудования и материалов с целью предотвращения их перепутывания и загрязнения.
4.12 Оборудование, обеспечивающее надлежащую защиту материалов (например, закрытая или изолированная система), может частично выходить за пределы производственного помещения.
4.13 При организации потоков материалов и персонала в зданиях или помещениях должно быть предусмотрено предотвращение перепутывания и загрязнения материалов и продукции.
4.14 Следует предусматривать отдельные зоны или другие адекватные решения для следующих операций:
- получения, идентификации, отбора проб и нахождения в карантине исходных материалов до выдачи разрешения на выпуск или отклонение;
- нахождения в карантине до выпуска или отклонения промежуточных продуктов или АФС;
- отбора проб промежуточных продуктов или АФС;
- хранения отклоненных материалов до принятия решения об их дальнейшем использовании (например, при возврате, переработке или уничтожении);
- хранения выпущенных материалов;
- выполнения технологических операций;
- выполнения операций по упаковке и маркировке;
- проведения лабораторных анализов.
4.15 Следует предусматривать необходимые помещения для подготовки персонала (мытье рук и пр.) и туалеты, обеспечить их горячей и холодной водой, мылом или другими моющими средствами, фенами для рук или одноразовыми полотенцами. Помещения для подготовки персонала и туалеты должны быть отделены от производственных зон (не иметь в них прямого выхода), но должны быть легкодоступными для персонала. При необходимости следует предусмотреть помещения для принятия душа и/или переодевания.
4.16 Как правило, лаборатории и помещения, в которых проводят анализы, должны быть расположены отдельно от производственных зон. Если проводимые в зонах технологические операции не влияют на результаты анализа, а работа лабораторий не оказывает отрицательного влияния на производство промежуточных продуктов и АФС, лаборатории (в частности, лаборатории внутрипроизводственного контроля) могут быть расположены в производственных зонах.
4.2 Инженерные системы
4.20 Следует проводить аттестацию инженерного оборудования и технологических сред (например, пара, газов, сжатого воздуха, систем отопления, вентиляции, кондиционирования и пр.), которые могут оказать влияние на качество продукции и обеспечить их контроль и техническое обслуживание. При несоответствии параметров установленным значениям следует принимать необходимые меры. На предприятии должен храниться соответствующий комплект документации.
4.21 При необходимости следует предусматривать системы вентиляции, очистки и вытяжки воздуха. При проектировании и мониторинге этих систем следует свести к минимуму риск прямого и перекрестного загрязнения и предусмотреть соответствующий контроль параметров на каждой технологической стадии (например, давления воздуха, наличия микроорганизмов (при необходимости), запыленности, влажности и температуры). Особое внимание следует уделить зонам, в которых АФС подвергаются воздействию окружающей среды.
4.22 В производственных помещениях с рециркуляцией воздуха следует предусмотреть меры по предотвращению риска прямого и перекрестного загрязнения.
4.23 Следует предусмотреть маркировку или иной метод идентификации стационарных систем трубопроводов (например, путем маркировки отдельных трубопроводов, обеспечением документации, применением систем с компьютерным управлением и контролем и пр.). Расположение системы трубопроводов не должно представлять опасность загрязнения промежуточных продуктов и АФС.
4.24 Канализационные трубы должны иметь соответствующие размеры и, при необходимости, иметь разрыв струи либо другие средства предотвращения обратного потока.
4.3 Подготовка воды
4.30 Вода, используемая при производстве АФС, должна соответствовать своему назначению, что должно быть подтверждено документально.
4.31 За исключением отдельно оговоренных случаев, качество воды, используемой в производстве, должно, как минимум, соответствовать требованиям действующих стандартов, предъявляемым к питьевой воде.
4.32 Если для обеспечения качества АФС характеристик питьевой воды недостаточно и необходимы более жесткие требования к химическим и/или микробиологическим характеристикам воды, должны быть разработаны требования к воде по физико-химическим свойствам, общему числу микроорганизмов, числу нежелательных микроорганизмов и/или содержанию эндотоксинов в воде.
4.33 Следует проводить аттестацию процесса подготовки воды определенного качества, используемой в технологическом процессе, и контролировать ее параметры с установлением необходимых пределов действия.
4.34 Если нестерильные АФС предназначены для дальнейшего производства стерильных лекарственных средств (или их производитель заявляет о пригодности АФС для этой цели), то вода, применяемая на конечных стадиях выделения и очистки, должна быть проверена на общее число микроорганизмов, число нежелательных микроорганизмов и уровень эндотоксинов.
4.4 Разделение зон
4.40 Для производства продукции с высокой сенсибилизирующей активностью (пенициллины или цефалоспорины и пр.) следует использовать специально выделенные производственные зоны, в состав которых могут входить помещения, вентиляционное и/или технологическое оборудование.
4.41 Специальные производственные площади должны быть выделены для производства продукции с инфицирующими свойствами, высокой фармакологической активностью или токсичностью (например, некоторых стероидов, цитотоксических противоопухолевых средств), за исключением случаев, когда методы инактивации и/или очистки оборудования прошли аттестацию (испытания) и выполняются в установленном порядке.
4.42 Следует предусмотреть меры по предотвращению перекрестного загрязнения при перемещении персонала, материалов и т.д. из одной зоны в другую.
4.43 Не допускается проводить в зданиях и/или на оборудовании, предназначенном для производства АФС, любые производственные операции (в т.ч. взвешивание, размол или упаковку) с высокотоксичными нефармацевтическими материалами, например, гербицидами и пестицидами. Обращение с высокотоксичными нефармацевтическими материалами и их хранение должны быть отделены от АФС.
4.5 Освещение
4.50 Для облегчения правильной эксплуатации, обслуживания и очистки помещений следует предусмотреть необходимый уровень их освещения.
4.6 Стоки и отходы
4.60 Стоки и отходы (например, твердые, жидкие или газообразные побочные продукты производства) должны своевременно и безопасно удаляться из зданий и окружающей их зоны с соблюдением санитарных норм. Контейнеры и/или трубопроводы для отходов должны иметь четкую маркировку.
4.7 Уборка, дезинфекция и техническое обслуживание
4.70 Здания и помещения, в которых производятся промежуточные продукты и АФС, должны соответствующим образом обслуживаться, ремонтироваться и содержаться в чистоте.
4.71 Для проведения уборки (обработки, дезинфекции) должны быть разработаны специальные инструкции с указанием используемого оборудования, материалов и графиков выполнения работ.
4.72 При необходимости для предотвращения загрязнения оборудования, сырья, упаковочных и печатных материалов, промежуточных продуктов и АФС следует разработать инструкции по борьбе с грызунами, использованию инсектицидов, фунгицидов, фумигирующих средств, а также чистящих и дезинфицирующих средств.
5.1 Требования к конструкции и монтажу
5.10 Оборудование, применяемое в производстве промежуточных продуктов и АФС, должно иметь соответствующую конструкцию и размеры и располагаться в месте, удобном для его использования, очистки, дезинфекции и технического обслуживания.
5.11 Поверхности оборудования, контактирующие с сырьем, промежуточными продуктами или АФС, не должны влиять на качество промежуточных продуктов и АФС и изменять их характеристики за пределы допустимых значений.
5.12 Технологическое оборудование должно использоваться только по назначению.
5.13 Основное оборудование и стационарные технологические линии (реакторы, контейнеры для хранения, технологические линии и пр.), используемые в производстве промежуточных продуктов и АФС, должны иметь соответствующую маркировку.
5.14 Не допускается контакт всех веществ, применяемых в технологическом оборудовании (смазок, нагревающих и охлаждающих жидкостей) с промежуточными продуктами и АФС, который может привести к изменению качества промежуточных продуктов и АФС за пределы установленных требований. Необходимо расследовать любые отклонения от этих требований, чтобы не допускать отрицательного влияния вспомогательных веществ на перерабатываемые материалы. По возможности следует использовать смазки и масла, предназначенные для пищевой промышленности.
5.15 Оборудование, по возможности, должно быть закрытым или герметичным. При использовании открытого оборудования необходимо принимать меры по снижению риска загрязнения до минимума.
5.16 Следует хранить действующий комплект документации на оборудование и установки (например, контрольно-измерительные приборы, вспомогательные системы и пр.).
5.2 Техническое обслуживание и очистка оборудования
5.20 Техническое обслуживание оборудования следует проводить в соответствии с утвержденными графиками и инструкциями, в которых должны быть указаны ответственные лица.
5.21 Инструкции по очистке оборудования и его допуску к последующему использованию в производстве промежуточных продуктов и АФС должны быть достаточно подробными и содержать следующие данные:
- определение ответственности за очистку оборудования;
- график проведения очистки и, при необходимости, дезинфекции;
- подробное описание методов и материалов, в т.ч. приготовления моющих средств;
- инструкции по разборке и сборке каждого узла оборудования (при необходимости);
- порядок удаления и уничтожения данных о предыдущей серии продукции;
- инструкции по предохранению чистого оборудования от загрязнения до начала его использования;
- порядок проверки чистоты оборудования непосредственно перед его использованием (при необходимости);
- максимально допустимое время после окончания производства до начала очистки оборудования (при необходимости).
5.22 Для предотвращения прямого и перекрестного загрязнений или переноса материалов, которые могут повлиять на соответствие качества промежуточных продуктов и АФС установленным требованиям, следует предусматривать надлежащее хранение, очистку оборудования и принадлежностей (при необходимости, дезинфекцию или стерилизацию).
5.23 Для оборудования, предназначенного для непрерывного производства или производства циклами серий одних и тех же промежуточных продуктов или АФС, следует предусмотреть его очистку через определенные интервалы времени для предотвращения накопления и переноса загрязнений (например, продуктов распада или недопустимой концентрации микроорганизмов).
5.24 Для неспециализированного оборудования между производством различных материалов следует предусмотреть его очистку с целью предупреждения перекрестного загрязнения.
5.25 Должны быть установлены допустимые предельные значения остатков загрязнений, а также методы и материалы, необходимые для очистки.
5.26 Проведение очистки оборудования должно быть документально оформлено. Оборудование должно быть промаркировано.
5.3 Калибровка (поверка)
5.30 Калибровку (поверку) контрольно-измерительного и аналитического оборудования (в т.ч. весов), имеющего критическое значение для обеспечения качества промежуточных продуктов или АФС, следует проводить в плановом порядке.
5.31 Калибровку (поверку) оборудования следует проводить с использованием стандартных образцов (эталонов), прослеживаемых до соответствующих эталонных образцов (если они существуют).
5.32 Протоколы проведения калибровок (поверок) должны сохраняться.
5.33 Данные о состоянии калибровки (поверки) критического оборудования должны находиться в доступном месте.
5.34 Не допускается использовать приборы, не соответствующие требованиям калибровки (поверки).
5.35 При обнаружении отклонений в работе контрольно-измерительного оборудования от установленных требований следует выяснить, могут ли они оказать влияние на качество промежуточных продуктов или АФС, произведенных с использованием этого оборудования после последнего проведения калибровки (поверки).
5.4 Системы с компьютерным управлением и контролем
5.40 Системы с компьютерным управлением и контролем должны быть аттестованы (испытаны) с учетом разнообразия, сложности и критичности использования компьютеров.
5.41 Пригодность систем с компьютерным управлением и контролем и программного обеспечения для выполнения поставленной задачи должна быть показана при их аттестации (испытаниях) в установленном и оснащенном состояниях.
5.42 Если программное обеспечение аттестовано до его получения, то не требуется проводить его проверку в аналогичном объеме. Если система с компьютерным управлением и контролем не была аттестована (испытана) при монтаже, то при наличии необходимой документации может быть проведена ее ретроспективная аттестация (испытания).
5.43 Следует предусмотреть защиту систем с компьютерным управлением и контролем от несанкционированного доступа или изменения данных, а также защиту от потери данных (например, при выключении компьютера).
Должна регистрироваться информация о любых изменениях данных, последнем вводе данных, о том, кем и когда они были сделаны.
5.44 Работа и обслуживание систем с компьютерным управлением и контролем должны проводиться в соответствии с инструкциями.
5.45 При вводе существенной информации вручную следует предусмотреть дополнительную проверку правильности ввода данных. Такая проверка может выполняться другим оператором либо самой системой.
5.46 Сбои в работе систем с компьютерным управлением и контролем, которые могут оказать влияние на качество промежуточных продуктов или АФС, на надежность записей или результатов испытаний, следует регистрировать и расследовать.
5.47 Внесение изменения в систему с компьютерным управлением и контролем должно проводиться в соответствии с методикой внесения изменений, оформляться документально и утверждаться ответственным лицом с проверкой внесения этих изменений. Следует хранить протоколы всех изменений и усовершенствований, внесенных в компьютер, программное обеспечение или любой другой критический элемент системы с компьютерным управлением и контролем.
5.48 Если сбои или отказы в работе системы могут привести к необратимой потере данных, следует предусмотреть резервную систему. Для всех систем с компьютерным управлением и контролем должны быть предусмотрены меры, гарантирующие защиту данных.
5.49 Допускается запись информации с помощью других средств.
6.1 Система документации и спецификации
6.10 Разработку, пересмотр, утверждение и распространение всех документов, относящихся к производству промежуточных продуктов или АФС, следует выполнять по специальным инструкциям (методикам). Эти документы могут быть оформлены как в письменном, так и в электронном виде.
6.11 Выпуск, пересмотр, замена и отмена всех документов должны контролироваться с сохранением сведений об их предыдущих версиях.
6.12 Следует организовать систему хранения всех необходимых документов (отчетов о развитии производства, объемах производства, замене оборудования и аттестации технологических процессов, протоколов обучения персонала, выпуска продукции, контроля качества и реализации продукции и пр.) и установить сроки хранения этих документов.
6.13 Вся документация о производстве, контроле качества и реализации продукции должна храниться не менее одного года после окончания срока годности данной серии. Для АФС с установленной датой повторного контроля срок хранения документов - не менее трех лет со дня полной реализации серии.
6.14 Записи в протоколы следует вносить в предназначенные для этого места сразу же после выполнения действий с указанием ответственного лица, сделавшего эти записи, так, чтобы их нельзя было удалить. Исправления в записях должны быть внесены таким образом, чтобы можно было прочесть первоначальную запись. Исправления должны содержать дату внесения изменений и подпись ответственного лица.
6.15 Оригиналы или копии протоколов, регистрирующие какие-либо действия, следует хранить в соответствующих подразделениях в течение всего срока хранения документации. Допускается хранить протоколы в других местах, если используется электронный или любой другой способ переноса информации.
6.16 Спецификации, инструкции, методики и протоколы могут храниться в виде оригиналов или заверенных копий (например, фотокопии, микрофильмы, микрофиши или другие точные воспроизведения оригинальных документов). При использовании микрофильмирования или записи в электронном виде следует иметь считывающее оборудование и средства для выполнения копий на жестких носителях.
6.17 Спецификации на сырье, промежуточные продукты (при необходимости), АФС, печатные и упаковочные материалы должны быть оформлены в письменном виде. Могут потребоваться спецификации для других материалов (технологических добавок, смазок и т.д.), используемых при производстве промежуточных продуктов или АФС, оказывающих решающее влияние на качество продукции. Критерии приемлемости для внутрипроизводственного контроля также должны быть оформлены в письменном виде.
6.18 Электронные подписи на документах должны быть идентифицированы и защищены.
6.2 Протокол очистки и использования оборудования
6.20 В протоколах использования, очистки, дезинфекции и/или стерилизации и обслуживания основного оборудования должны быть указаны дата, время, наименование и номер каждой серии произведенной на нем продукции, а также информация о лице, проводившем очистку и обслуживание этого оборудования.
6.21 Не требуется составление отдельных протоколов очистки и использования оборудования в случае, если оно специально предназначено для производства одного промежуточного продукта или АФС, и серии этого промежуточного продукта или АФС производятся в прослеживаемой последовательности. При использовании оборудования специального назначения протоколы очистки, обслуживания и использования могут быть частью протокола на серию продукции или быть оформлены в виде отдельных документов.
6.3 Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы для АФС
6.30 Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы, используемые для производства АФС, должны храниться в установленном порядке и содержать следующие данные:
- наименование производителя, содержание и количество каждой поставки каждой партии сырья, промежуточных продуктов или печатных и упаковочных материалов для производства АФС, наименование поставщика, шифр(ы) или другой идентификационный номер поставщика (если известны), номер, присвоенный при поступлении, и дата поступления;
- результаты проведенных испытаний или исследований и выводы по ним;
- протоколы, прослеживающие использование материалов;
- протокол о соответствии материалов, используемых для маркировки и упаковки АФС, требованиям, установленным в спецификациях и
- принятое решение относительно отклоненного сырья, промежуточных продуктов или печатных и упаковочных материалов, используемых в производстве АФС.
6.31 Следует хранить утвержденные образцы этикеток для сравнения с ними выпускаемых этикеток.
6.4 Промышленные регламенты
6.40 С целью обеспечения однообразия и повторяемости от серии к серии для каждого промежуточного продукта и АФС должны быть разработаны промышленные регламенты, подписанные одним лицом с указанием даты, проверенные и подписанные другим независимым лицом со стороны отдела(ов) качества с указанием даты.
6.41 Промышленные регламенты должны содержать следующие данные:
- наименование производимого промежуточного продукта или АФС и ссылку на соответствующий документ (если это возможно);
- полный перечень сырья и промежуточных продуктов согласно их наименованиям или кодам, достаточным для идентификации всех их показателей качества;
- количество или соотношение используемых видов сырья или промежуточного продукта, в т.ч. единицу его измерения. Если это количество не является фиксированной величиной, то в протокол должен быть включен расчет для каждого размера серии или производительности. При наличии обоснования в протокол следует включать допустимые отклонения значений;
- место производства и основное оборудование;
- подробные технологические инструкции, включающие в себя:
- последовательность операций;
- допустимые пределы изменения технологических параметров;
- инструкции по отбору проб и проведению внутрипроизводственного контроля с указанием критериев приемлемости (при необходимости);
- ограничения по времени завершения отдельных технологических стадий и/или всего технологического процесса;
- предполагаемый выход продукта (с указанием наибольшего и наименьшего значений) для соответствующих стадий технологического процесса или отрезков времени;
- особые указания и меры предосторожности или ссылки на них (при необходимости);
- инструкции по хранению промежуточного продукта или АФС, печатных и упаковочных материалов и особых условий хранения с указанием сроков годности.
6.5 Протоколы на серию продукции (протоколы производства и контроля качества серии продукции)
6.50 Для каждого промежуточного продукта и АФС составляют протоколы серии продукции, включающие в себя полную информацию о производстве и контроле качества каждой серии. До выпуска продукции протокол на серию должен быть проверен с целью подтверждения содержащихся в нем данных и их соответствия промышленному регламенту. Если протокол на серию продукции относится к части промышленного регламента, в протоколе должна быть ссылка на соответствующий промышленный регламент.
6.51 Протоколы на серию продукции должны быть пронумерованы с использованием уникального номера серии или идентификационного кода и подписаны с указанием даты. При непрерывном производстве до присвоения окончательного номера серии в качестве идентификационного кода допускается использовать шифр продукта с указанием даты и времени.
6.52 В протоколах на серию продукции после завершения каждой важной технологической стадии следует указывать:
- дату и время, при необходимости;
- основное используемое оборудование (например, реакторы, сушильные аппараты, мельницы и т.д.);
- данные о каждой серии (в т.ч. взвешивание, отмеривание и номера серий сырья, промежуточных продуктов или любых других материалов, использованных в производстве);
- фактические данные критических технологических параметров;
- данные о всех отобранных пробах;
- подписи непосредственных исполнителей и лиц, проверяющих или контролирующих каждую критическую операцию в данном процессе;
- результаты внутрипроизводственного и лабораторного контроля;
- фактический выход продукции на соответствующих стадиях или на определенный момент времени;
- данные об упаковке и маркировке промежуточного продукта или АФС;
- образцы маркировки АФС или промежуточных продуктов, если их поставляют в готовом виде;
- любые отклонения, их оценку, результаты расследования (при необходимости) или ссылку на проведенное расследование (если оно хранится в виде отдельного протокола);
- результаты контроля при выпуске продукции.
6.53 Следует разработать инструкции по расследованию критических отклонений и несоответствия серии промежуточных материалов или АФС требованиям спецификаций. В это расследование следует включать и другие серии продукции, которые могут быть связаны с данным несоответствием или отклонением.
6.6 Протоколы лабораторного контроля
6.60 В протоколах лабораторного контроля должна быть полная информация о ходе всех испытаний, проведенных для подтверждения соответствия требованиям нормативной документации, в т.ч. данные наблюдений и анализов:
- описание полученных проб, в т.ч. наименование материала или место отбора пробы, номер серии или другой отличительный код, дата взятия образца, количество и дата поступления этой пробы в лабораторию;
- указание и/или ссылка на каждый использованный метод контроля;
- указание массы или размеров пробы для каждого испытания согласно принятой методике; данные или ссылка на приготовление и проведение испытания образцов сравнения, реактивов и стандартных растворов;
- необработанные в процессе каждого испытания данные в дополнение к графикам, таблицам и спектрам, полученным с использованием лабораторного оборудования с нумерацией (кодом), позволяющей определить испытуемое вещество и серию;
- вычисления с указанием единиц измерения, коэффициентов перевода и коэффициентов эквивалентности;
- результаты контроля и их сравнения с установленными критериями приемлемости;
- подпись ответственного лица, проводившего каждый вид контроля, и дату(ы) его проведения и
- дата и подпись второго лица, заверяющая точность, полноту и соответствие установленным требованиям.
6.61 Следует также составлять протоколы с указанием:
- любых изменений в принятых методах контроля;
- периодической калибровки (поверки) лабораторных приборов, оборудования и записывающих устройств;
- испытаний на стабильность АФС;
- результатов исследований, не удовлетворяющих требованиям спецификации.
6.7 Рассмотрение протоколов на серию продукции
6.70 Для определения соответствия промежуточного продукта или АФС установленным требованиям до выпуска или распространения данной серии продукции следует разработать инструкции по рассмотрению и утверждению (согласованию) протоколов на серию продукции и проведению лабораторного контроля качества продукции, в т.ч. упаковку и маркировку.
6.71 Протоколы на серию и лабораторные протоколы контроля критических технологических стадий до выпуска и реализации серии АФС должны быть рассмотрены и утверждены отделом(ами) контроля качества. Протоколы внутрипроизводственного и лабораторного контроля некритических технологических стадий могут быть рассмотрены квалифицированным производственным персоналом или другими отделами в соответствии с инструкциями, согласованными с отделом(ами) качества.
6.72 Все отклонения от спецификаций и расследования этих случаев должны быть рассмотрены до выпуска серии в реализацию.
6.73 Служба (отделы) контроля качества может передать ответственность и право выдачи разрешения на выпуск промежуточных продуктов производственному отделу (службе), за исключением случаев, когда промежуточные продукты поставляют за пределы зоны их контроля предприятием-производителем.
7.1 Общий контроль
7.10 Порядок получения, идентификации, карантинного хранения, обращения, отбора проб, проведения контроля и выдачи разрешения на использование или отклонение материалов должен быть установлен в соответствующих инструкциях.
7.11 У производителей промежуточных продуктов и/или АФС должна быть разработана и внедрена система оценки поставщиков критических материалов.
7.12 Поставка материалов должна осуществляться поставщиками, утвержденными отделом(ами) качества, в соответствии с согласованными спецификациями.
7.13 Если поставщик критического материала не является производителем данного материала, производитель промежуточных продуктов и/или АФС должен иметь информацию о наименовании и адресе этого производителя.
7.14 Замена поставщика критического сырья должна проводиться в соответствии с процедурой, изложенной в разделе 13.
7.2 Приемка и карантин
7.20 Перед приемкой и при получении материалов на каждой упаковке или группе упаковок должны быть визуально проверены правильность маркировки (в т.ч. соответствие названия, используемого поставщиком и заказчиком, если они отличаются), сохранность упаковок, наличие поврежденных печатей, следов вскрытия и загрязнения упаковок. Материалы должны находиться на карантинном хранении до проведения отбора проб, их анализа (контроля) и получения разрешения на их использование.
7.21 До смешивания поступивших материалов с имеющимися запасами (например, с растворителями или запасами, находящимися в хранилищах) следует установить их подлинность, при необходимости - провести контроль и получить разрешение на их использование. Для предотвращения неправильного размещения поступивших материалов среди уже хранящихся материалов должны быть разработаны специальные инструкции.
7.22 Следует исключить возможность перекрестного загрязнения нерасфасованной продукции, если она поставляется в емкостях, не предназначенных специально для нее. В качестве доказательства этого могут использоваться:
- паспорт очистки;
- контроль на наличие следов примесей;
- аудит поставщика.
7.23 Большие емкости для хранения и соответствующие коллекторы, линии по загрузке и выгрузке должны иметь соответствующую маркировку.
7.24 Каждая емкость или группа емкостей (серий) материалов должна иметь четкую маркировку (шифр, номер серии или поставки, контракта, товарной накладной). Эти номера должны использоваться при обозначении расположения каждой серии. Должна быть разработана и внедрена система обозначения статуса каждой серии.
7.3 Отбор проб и проведение испытаний поступивших материалов
7.30 Для подтверждения подлинности каждой серии материалов (за исключением материалов, указанных в 7.32) следует провести хотя бы одно испытание. Вместо проведения последующих испытаний можно использовать аналитический паспорт поставщика при условии, что на предприятии производителя действует система оценки поставщиков.
7.31 Система оценки поставщиков должна включать в себя доказательства (например, данные о качестве предыдущих поставок) того, что производитель может стабильно поставлять материалы, соответствующие требованиям спецификации. Прежде чем упростить процедуру собственных проверок, следует провести полный анализ не менее трех серий материала. Полный анализ должен проводиться через определенные интервалы времени и сравниваться с аналитическим паспортом. Достоверность аналитического паспорта необходимо регулярно проверять.
7.32 Не требуется проведение контроля технологических добавок, опасного или высокотоксичного сырья, других специальных материалов или материалов, передаваемых в другое подразделение под контролем заказчика, при наличии аналитического паспорта производителя, подтверждающего соответствие этого сырья установленным требованиям. Идентификацию этих материалов проводят путем визуального осмотра упаковок, этикеток и регистрации номеров серий. Если контроль таких материалов на месте не проводится, то это должно быть обосновано и оформлено документально.
7.33 Пробы должны быть репрезентативной выборкой серии материала, из которой они отобраны. В методике отбора проб следует указывать число контейнеров, из которых должны быть взяты пробы, из какой части контейнера и в каком количестве их следует отбирать. Число контейнеров, из которых следует отобрать пробы, и количество проб должны быть установлены в плане отбора проб. Следует учитывать критичность и стабильность свойств материала, информацию о качестве предыдущих поставок и количестве материала, необходимого для проведения анализа.
7.34 Отбор проб следует проводить в специально отведенном месте. Методика отбора проб должна предусматривать предотвращение загрязнения материала, из которого отбираются пробы, и других материалов.
7.35 Упаковки, из которых отбирают пробы, должны быть аккуратно вскрыты и закрыты. На упаковках должна быть нанесена маркировка, показывающая, что из этой упаковки были взяты пробы.
7.4 Хранение
7.40 При обращении с материалами и их хранении не допускаются ухудшение их качества, загрязнение и перекрестное загрязнение.
7.41 Материалы, хранящиеся в картонных барабанах, мешках или коробках, должны быть расположены таким образом, чтобы было удобно проводить уборку и проверку. Не допускается складирование материалов на полу.
7.42 Период и условия хранения материалов не должны отрицательно влиять на их качество. Как правило, материалы, поступившие первыми, следует использовать в первую очередь.
7.43 Некоторые материалы можно хранить вне помещений при условии надлежащей упаковки, читаемой маркировки и выполнения необходимой очистки упаковок перед их вскрытием и использованием.
7.44 Отклоненные материалы должны иметь соответствующую маркировку и помещаться на карантин (в изолятор брака) для исключения их несанкционированного использования в производстве.
7.5 Повторный контроль
7.50 При необходимости, для определения пригодности материалов (например, после длительного хранения или в результате воздействия температуры или влажности) следует проводить повторный контроль.
8.1 Технологические операции
8.10 Условия взвешивания и отмеривания сырья для производства промежуточных продуктов и АФС не должны оказывать влияния на их пригодность. Устройства для взвешивания и отмеривания должны быть необходимой точности.
8.11 Если материал разделяется на части для последующего использования в производственных операциях, то он должен помещаться в специальную упаковку, маркировка которой содержит следующую информацию:
- наименование материала и/или код;
- номер, присвоенный при получении, или контрольный номер;
- массу или меру материала в новой упаковке и
- дату повторного контроля, при необходимости.
8.12 Выполнение критических операций по взвешиванию, отмериванию или разделению материала на порции должно быть заверено или проконтролировано эквивалентным образом. До начала работы производственный персонал должен убедиться в том, что все полученные материалы соответствуют данным, указанным в протоколе серии для производства данного промежуточного продукта или АФС.
8.13 Выполнение других критических действий должно быть заверено или проконтролировано эквивалентным образом.
8.14 На определенных стадиях технологического процесса необходимо сравнивать фактический выход продукции (материалов) с ожидаемым. Ожидаемый выход и допустимые отклонения должны быть определены на основании результатов лабораторных, опытных или производственных испытаний, проведенных предварительно. Отклонения от ожидаемого выхода, связанные с критическими технологическими стадиями, должны быть расследованы для определения их возможного влияния на качество серии продукции.
8.15 Все отклонения должны быть оформлены документально с указанием причин. Любое критическое отклонение должно быть расследовано.
8.16 Назначение основных единиц оборудования должно быть указано на самом оборудовании или отражено в соответствующей документации, системе с компьютерным управлением и контролем или другим образом.
8.17 Следует организовать контроль материалов, предназначенных для повторного использования или переработки во избежание их несанкционированного использования.
8.2 Ограничения на время выполнения операций
8.20 Промышленным регламентом могут быть установлены ограничения на время выполнения операций (6.41). Отклонения от этих ограничений следует оформлять документально и проводить их оценку. Такие ограничения могут не потребоваться при проведении технологического процесса до достижения установленных значений параметров (например, доводка рН, гидрогенизация, сушка до значения, установленного спецификацией), так как в этом случае завершение реакций или стадий производства определяется при внутрипроизводственном отборе проб и контроле.
8.21 Условия хранения промежуточных продуктов должны обеспечивать их дальнейшую пригодность.
8.3 Внутрипроизводственный отбор проб и контроль
8.30 Для контроля и выполнения технологических стадий, которые влияют на показатели качества промежуточных продуктов и АФС, должны быть разработаны инструкции. Порядок проведения внутрипроизводственного контроля и критерии приемлемости определяют на основании информации, полученной при разработке технологического процесса или по предыдущему опыту производства.
8.31 Критерии приемлемости, вид и объем испытаний могут зависеть от вида производимых промежуточных продуктов или АФС, проводимой реакции или технологической стадии и степени влияния этого процесса на качество продукции. На ранних технологических стадиях допускается проводить менее строгий внутрипроизводственный контроль; на более поздних технологических стадиях (например, на стадиях выделения и очистки) - контроль может быть более жестким.
8.32 Порядок проведения внутрипроизводственного контроля на критических стадиях должен включать в себя контрольные точки и методы контроля, быть оформлен в письменном виде и согласован с отделом контроля качества.
8.33 Внутрипроизводственный контроль может проводить персонал производственного подразделения, имеющий необходимую квалификацию, а наладку технологического процесса допускается проводить без разрешения службы контроля качества, если она осуществляется в установленных допустимых пределах, согласованных с отделом контроля качества. Результаты такого контроля должны быть документально оформлены в полном объеме и входить в протокол на серию продукции.
8.34 Порядок отбора проб промежуточных материалов, продуктов и АФС должен быть приведен в документально оформленных и утвержденных в установленном порядке методиках. Планы и методики отбора проб должны быть обоснованными.
8.35 Внутрипроизводственный отбор проб должен проводиться с использованием методик, предусматривающих предотвращение загрязнения материалов и других промежуточных продуктов или АФС. Применяемые методы должны обеспечивать целостность проб после их отбора.
8.36 Если при проверке и/или наладке технологического процесса результаты внутрипроизводственного контроля окажутся не соответствующими требованиям спецификаций, то проведение анализа причин отклонений, как правило, не требуется.
8.4 Смешанные серии промежуточных продуктов или АФС
8.40 В данном руководстве смешивание материалов определяется как процесс объединения материалов в рамках одной спецификации для производства однородного промежуточного продукта или АФС. Внутрипроизводственное смешивание фракций одной серии (например, объединение нескольких загрузок центрифуги из одной кристаллизационной серии или объединение фракций из нескольких серий для дальнейшего производства) рассматривается как часть технологического процесса и не считается смешиванием.
8.41 Не допускается смешивание серии, не удовлетворяющей требованиям спецификации, с другими сериями. Каждая серия, вносимая в смесь, должна быть произведена согласно установленным требованиям, индивидуально испытана, и ее соответствие спецификации должно быть подтверждено до смешивания.
8.42 Как правило, смешиваются:
- малые серии для увеличения ее размера;
- остатки (т.е. относительно малые количества выделенного материала) из серий одного и того же промежуточного продукта или АФС для формирования одной серии.
При необходимости допускаются и другие случаи смешивания материалов.
8.43 Процессы смешивания следует контролировать и оформлять в документальном виде, а смешанная серия должна быть, при необходимости, испытана на соответствие установленным требованиям.
8.44 В протоколе на серию, полученную после смешивания, должен прослеживаться процесс производства индивидуальных серий, формирующих эту смешанную серию.
8.45 Если физические характеристики АФС являются критическими (например, АФС предназначена для использования в твердых дозированных формах для принятия внутрь или в суспензиях), то операции по смешиванию должны быть аттестованы для подтверждения однородности окончательной серии. Аттестация должна включать в себя испытание критических параметров (распределение частиц по размеру, объемная плотность при свободной засыпке и удельная плотность при уплотнении), на которые может повлиять процесс смешивания.
8.46 Если смешивание материалов может оказать неблагоприятное воздействие на стабильность продукции, то следует провести испытания стабильности окончательной серии.
8.47 Срок годности или дата проведения повторного контроля смешанной серии должны быть основаны на дате производства самой старой фракции или серии в смеси.
8.5 Контроль загрязнений
8.50 Допускается переносить остатки материалов в последующие серии того же промежуточного продукта или АФС при наличии соответствующего контроля. К ним относятся остатки, прилипающие к стенкам измельчителя, слой влажных кристаллов, остающийся в центрифуге после разгрузки, и неполное удаление жидкостей или кристаллов из технологической емкости после передачи материала на следующую технологическую стадию. Такой перенос не должен приводить к переносу продуктов распада или микробного загрязнения, которые могут оказать отрицательное влияние на чистоту АФС.
8.51 При выполнении технологических операций не следует допускать загрязнения промежуточных продуктов или АФС другими материалами.
8.52 После проведения очистки оборудования следует принять меры предосторожности, чтобы избежать загрязнения АФС.
9.1 Общие положения
9.10 Следует разработать и документально оформить методики на получение, идентификацию, карантинное хранение, отбор проб, проведение исследования и/или испытания и выдачу разрешения на использование, а также на обращение с упаковочными и печатными материалами.
9.11 Материалы для упаковки и маркировки должны соответствовать установленным требованиям. Материалы, не соответствующие этим требованиям, должны отклоняться для предотвращения их использования в операциях, для которых они непригодны.
9.12 Следует хранить протоколы на каждую поставку печатных и упаковочных материалов, содержащие информацию о поставке, осмотре или контроле этого материала, а также заключение о разрешении его использования или отклонения.
9.2 Упаковочные материалы
9.20 Упаковка должна обеспечивать надлежащую защиту промежуточного продукта или АФС от порчи или загрязнения, которые могут произойти в процессе их транспортирования или хранения.
9.21 Упаковки должны быть чистыми и, если это обусловлено видом промежуточного продукта или АФС, должны пройти обработку (дезинфекцию) для обеспечения их пригодности для дальнейшего использования. Упакованные материалы не должны вступать в химические реакции и обладать аддитивными или абсорбирующими свойствами, чтобы не допустить изменения качества промежуточного продукта или АФС за установленные пределы.
9.22 При повторном использовании упаковки должны быть очищены в соответствии с утвержденными инструкциями, а все предыдущие этикетки удалены.
9.3 Выпуск и контроль печатных материалов
9.30 Доступ к местам хранения печатных материалов должен быть разрешен только лицам, имеющим соответствующие полномочия.
9.31 Порядок определения соответствия между количеством отпущенного, использованного и возвращенного печатного материала и оценки расхождений между количеством маркированных упаковок и выданных этикеток должен быть указан в инструкции. Факты несоответствия должны расследоваться, а результаты расследования должны быть утверждены отделом контроля качества.
9.32 Весь неиспользованный печатный материал, содержащий номер серии или другую печатную информацию о серии, должен быть уничтожен. Возвращенный печатный материал должен храниться таким образом, чтобы предотвратить его перепутывание и обеспечить надлежащую идентификацию.
9.33 Вышедший из употребления и устаревший печатный материал должен быть уничтожен.
9.34 Следует организовать контроль за работой устройств, предназначенных для печатания этикеток для упаковочных операций, чтобы гарантировать соответствие всех оттисков протоколу на серию продукции.
9.35 Следует организовать тщательный контроль этикеток, отпечатанных для серии, на их соответствие данной серии и установленным требованиям. Результат контроля должен быть оформлен документально.
9.36 Образец используемой отпечатанной этикетки должен быть включен в протокол серии продукции.
9.4 Операции по упаковке и маркировке
9.40 Следует разработать и документально оформить инструкции по использованию упаковочных и печатных материалов.
9.41 Следует разработать инструкции по выполнению маркировки, исключающие перепутывание печатных материалов. Операции по маркировке различных промежуточных продуктов или АФС должны быть разграничены физически или пространственно.
9.42 Этикетки, используемые на упаковках промежуточных продуктов или АФС, должны содержать наименование или идентификационный код, номер серии продукта и условия хранения (в том случае, когда эта информация является критической для обеспечения качества промежуточного продукта или АФС).
9.43 Если промежуточный продукт или АФС предназначены для передачи за пределы зоны действия системы контроля их производителем, то маркировка также должна содержать наименование и адрес предприятия-производителя, количество содержимого, специальные условия транспортирования и любые другие требования, предусмотренные нормативной документацией. Если для промежуточных продуктов или АФС установлен срок годности, то он должен быть указан на маркировке и в аналитическом паспорте. Для промежуточных материалов или АФС, прошедших повторный контроль, дата повторного испытания должна быть указана на маркировке и в аналитическом паспорте.
9.44 Упаковочное и печатное оборудование должно быть проверено непосредственно перед использованием для гарантии того, что все материалы, не нужные для последующей упаковки, были удалены. Эта проверка должна быть указана в протоколе на серию продукции, журнале оборудования или другой документации.
9.45 В состав операций по упаковке входит проверка правильности маркировки первичных и вторичных упаковок промежуточных продуктов или АФС. Результаты этих проверок должны быть указаны в протоколах на серию продукции или контроль качества продукции.
9.46 Промежуточные продукты или АФС, отправляемые за пределы зоны контроля производителя, должны быть опечатаны таким образом, чтобы при повреждении или отсутствии печати получатель обратил внимание на возможность замены содержимого.
10.1 Хранение на складе
10.10 В помещениях для хранения материалов должны быть предусмотрены соответствующие условия (например, поддерживание заданной температуры и влажности, при необходимости). Следует вести протоколы условий хранения, если они являются критическими для сохранения свойств материалов.
10.11 Во избежание случайного или несанкционированного использования находящихся в карантине, отклоненных, возвращенных или отозванных материалов должны быть выделены отдельные зоны для их временного хранения до принятия решения об их дальнейшем использовании (если другое не указано в документации).
10.2 Реализация
10.20 Реализация АФС и промежуточных продуктов третьим сторонам допускается только после получения разрешения отдела контроля качества на их выпуск. В условиях карантина АФС и промежуточные продукты могут быть переведены в другое подразделение при наличии разрешения отдела контроля качества, соответствующей документации и организации надлежащего контроля.
10.21 Транспортирование АФС и промежуточных продуктов не должно оказывать влияния на их качество.
10.22 Особые условия транспортирования и хранения АФС и промежуточных продуктов должны быть указаны на этикетке.
10.23 Производитель должен убедиться в том, что подрядчик, осуществляющий транспортирование АФС и промежуточных продуктов по контракту, знает и соблюдает требуемые условия транспортирования и хранения.
10.24 Для обеспечения возможности быстрого отзыва каждой серии продукции должна быть организована система отслеживания за ее реализацией.
11.1 Общий контроль
11.10 Служба контроля качества должна иметь в своем распоряжении необходимые лабораторные помещения и оборудование.
11.11 Следует разработать и документально оформить методики отбора проб, проведения испытаний, выдачи разрешения на использование или отклонение материалов, регистрации и хранения данных, полученных в лаборатории. Ведение протоколов и отчетов должно соответствовать требованиям 6.6.
11.12 Все спецификации, планы отбора проб и методики проведения испытаний должны быть научно обоснованными и гарантировать, что сырье, АФС и промежуточные продукты, а также печатные и упаковочные материалы соответствуют установленным стандартам качества и/или чистоты. Спецификации и методики испытаний должны соответствовать требованиям, установленным при государственной регистрации. Допускается использовать спецификации, не входящие в регистрационное досье. Спецификации, планы отбора проб и методики испытаний, в т.ч. изменения к ним, должны быть разработаны соответствующим подразделением, рассмотрены и согласованы со службой (отделом) контроля качества.
11.13 Спецификации для АФС должны быть разработаны в соответствии с действующими нормами и технологическим процессом и включать в себя контроль за примесями (например, органическими и неорганическими примесями и остаточным содержанием растворителей). Если для АФС предусмотрены требования к микробиологической чистоте, то в спецификации должны быть установлены допустимые пределы для общего числа микроорганизмов и нежелательных микроорганизмов. Если для АФС предусмотрены требования к апирогенности, то в спецификации должны быть установлены допустимые пределы к уровню эндотоксинов.
11.14 Все операции контроля в лабораториях должны проводиться в соответствии с утвержденными методиками (инструкциями) и оформляться в письменном виде непосредственно после их проведения. Любые отклонения от указанных методик должны быть обоснованы.
11.15 Любые результаты, выходящие за рамки спецификаций, должны быть расследованы и оформлены в соответствии с принятой методикой, предусматривающей проведение анализа данных, оценку существенной проблемы, определение путей решения этой проблемы и формулирование выводов из проведенного расследования. После получения результатов, выходящих за рамки спецификаций, повторные отборы проб и/или повторные испытания должны проводиться в соответствии с принятыми методиками.
11.16 Реактивы и стандартные растворы должны приготавливаться и маркироваться в соответствии с принятой методикой. Для аналитических реактивов и растворов следует применять маркировку "Использовать до".
11.17 Для производства АФС необходимо иметь первичные стандартные образцы и документацию о происхождении каждого стандартного образца. Данные о хранении и использовании каждого первичного стандартного образца согласно рекомендациям поставщика должны быть приведены в соответствующих протоколах. Первичные стандартные образцы, полученные из официально признанных источников и хранящиеся в соответствии с рекомендациями поставщика, используют без проведения испытаний.
11.18 При невозможности получения первичных стандартных образцов из официально признанных источников следует разработать внутренние стандартные образцы производителя. Для установления подлинности и чистоты первичного стандартного образца требуется проведение соответствующего контроля. Проведение контроля должно быть оформлено документально.
11.19 Следует разработать порядок изготовления, установления, идентификации, испытаний, утверждения и хранения вторичных стандартных образцов. Пригодность каждой серии вторичного стандартного образца должна быть определена до момента первого использования путем его сравнения с первичным стандартным образцом. Каждая серия вторичного стандартного образца должна периодически проходить проверку (аттестацию) в соответствии с утвержденной методикой.
11.2 Проведение испытаний промежуточных продуктов и АФС
11.20 Для подтверждения соответствия каждой серии промежуточного продукта и АФС требованиям спецификаций должны быть проведены необходимые лабораторные испытания.
11.21 Для каждой АФС должен быть определен состав примесей (идентифицированных и неидентифицированных), присутствующих в обычной серии, произведенной в ходе конкретного контролируемого технологического процесса. Описание примесей должно включать в себя их идентификацию или какие-либо качественные аналитические параметры, позволяющие ее идентифицировать (например, время удерживания), диапазон содержания каждой выявленной примеси и их классификацию (неорганическая, органическая, растворитель). Состав примесей обычно зависит от технологического процесса и природы АФС. Для АФС, получаемых из растительного сырья или из тканей животных, определение состава примесей обычно не требуется. Требования к АФС, получаемым биотехнологическим путем, должны быть установлены в соответствующей нормативной документации.
11.22 Для выявления изменений в АФС, происходящих вследствие изменений в сырье, оборудовании, рабочих параметрах или технологическом процессе, состав примесей периодически следует сравнивать с составом примесей, указанных в документации, оформляемой при государственной регистрации, или с данными, полученными ранее.
11.23 Следует проводить контроль микробной чистоты каждой серии промежуточного продукта и АФС в соответствии с установленными требованиями.
11.3 Аттестация аналитических методик (см. раздел 12)
11.4 Аналитические паспорта
11.40 На каждую серию промежуточного продукта и/или АФС должен выдаваться подлинный аналитический паспорт.
11.41 Аналитический паспорт должен содержать информацию о наименовании промежуточного продукта или АФС, в т.ч. о его чистоте, номере серии и дате выпуска. Для промежуточных продуктов и АФС с установленным сроком годности последний должен быть указан в маркировке и аналитическом паспорте. Для промежуточных продуктов или АФС с датой повторного контроля эта дата также должна быть указана в маркировке и/или аналитическом паспорте.
11.42 В аналитическом паспорте должны быть перечислены все испытания, проведенные в соответствии с нормативными документами или требованиями заказчика, в т.ч. критерии приемлемости и полученные количественные результаты (при их наличии).
11.43 В аналитическом паспорте должна быть указана дата и подпись ответственного лица отдела контроля качества, а также наименование, адрес и телефон первичного предприятия-производителя. Если анализ был проведен на вторичном предприятии-упаковщике или производителе, аналитический паспорт должен содержать наименование, адрес, телефон вторичного предприятия-упаковщика или производителя и наименование первичного производителя.
11.44 Новые аналитические паспорта, выпускаемые вторичным предприятием-упаковщиком или производителем, поставщиками или лицами, действующими от их имени, должны содержать наименование, адрес и телефон лаборатории, проводившей эти анализы. В них также должны быть указаны наименование, адрес первичного производителя и оригинал паспорта на эту серию продукции с приложением его копии.
11.5 Контроль стабильности АФС
11.50 Следует разработать и документально оформить программу непрерывного контроля стабильности АФС, результаты которого должны использоваться для подтверждения условий хранения при пересмотре срока годности или срока проведения повторного контроля.
11.51 Методики испытаний на стабильность должны быть аттестованы.
11.52 Пробы для контроля стабильности следует хранить в упаковках, имитирующих коммерческие упаковки. Например, для АФС, поставляемых в мешках, помещенных в картонные барабаны, пробы для контроля стабильности могут быть упакованы в мешки из того же материала, помещенные в барабаны, меньшие по размеру и изготовленные из похожего или такого же материала, как и для АФС, поступающих в реализацию.
11.53 Для подтверждения даты проведения повторного контроля или срока годности АФС первые три реализуемые серии, как правило, должны быть испытаны на стабильность. Если результаты предыдущих испытаний показывают, что данная серия АФС будет сохранять стабильность еще не менее двух лет, допускается использовать менее трех серий.
11.54 В программу контроля стабильности ежегодно должна включаться, по крайней мере, хотя бы одна серия производимой АФС (за исключением случаев, когда в этот год ее производство не осуществлялось).
11.55 Для АФС с коротким сроком годности испытания следует проводить чаще. Например, для биотехнологических/биологических и других АФС со сроком хранения один год и менее пробы на стабильность следует отбирать и испытывать ежемесячно в течение первых 3 мес и затем с интервалом в 3 мес.
При наличии данных, подтверждающих сохранение стабильности АФС, может быть рассмотрен вопрос об увеличении периодичности испытаний (например, проведение испытаний через 9 мес).
11.56 Условия хранения при проведении испытаний на стабильность должны, по возможности, соответствовать требованиям нормативной документации к проведению этих испытаний.
11.6 Срок годности и дата повторного контроля
11.60 Если промежуточный продукт предназначен для перемещения за пределы зоны действия системы управления качеством производителя и для этого продукта установлен срок годности или дата проведения повторного контроля, то следует иметь данные по его стабильности (например, опубликованную информацию, результаты испытаний).
11.61 Срок годности АФС и дата повторного контроля должны быть основаны на результатах анализа данных, полученных при испытаниях на стабильность. Обычно для АФС используют дату повторного контроля, а не дату срока годности.
11.62 Определение предварительного срока годности или даты повторного контроля АФС может основываться на данных производства опытных серий в следующих случаях:
1) при производстве опытных серий использовались методы, моделирующие технологический процесс при промышленном производстве;
2) качество АФС соответствует качеству продукции, которая будет производиться в промышленном масштабе.
11.63 Для проведения повторного контроля должен использоваться образец, репрезентативный для этой серии.
11.7 Архивные образцы
11.70 Упаковка и хранение архивных образцов предназначены для возможной оценки качества серий АФС в будущем, а не для проведения испытаний этих образцов на стабильность.
11.71 Маркированные архивные образцы из каждой серии АФС следует сохранять не менее одного года после истечения срока годности данной серии, определенного производителем, или в течение трех лет после даты реализации этой серии, в зависимости от того, какой срок больше. Для АФС с датой повторного контроля аналогичные архивные образцы следует хранить в течение трех лет после того, как эта серия была полностью реализована производителем.
11.72 Архивные образцы должны храниться в такой же или в эквивалентной упаковке, используемой для АФС, либо в упаковке, более защищенной, чем упаковка, в которой реализуется эта продукция. Количество архивных образцов должно быть достаточным для проведения не менее двух полных анализов в соответствии с требованиями Фармакопеи, либо, в случае отсутствия соответствующей фармакопейной статьи, двух полных анализов согласно спецификации.
12.1 Политика проведения аттестации (испытаний)
12.10 Общая политика предприятия, цели и подход к процессам аттестации, в т.ч. к аттестации технологических процессов, методов очистки, аналитических методов, методов проведения внутрипроизводственного контроля, систем с компьютерным управлением и контролем и лиц, ответственных за разработку, рассмотрение, согласование и оформление каждой стадии аттестации (испытаний), должны быть оформлены в письменном виде.
12.11 Как правило, в ходе разработки процесса (или по имеющимся данным) должны быть определены критические параметры (характеристики) и пределы их изменений, необходимые для обеспечения надежной работы. К таким параметрам относятся:
- критические характеристики, которые должны быть указаны для данного продукта (АФС);
- технологические параметры, которые могут повлиять на критические характеристики качества АФС;
- предельные изменения каждого критического параметра процесса, которые будут использоваться в ходе текущего производственного контроля.
12.12 Операции, которые считаются критическими для качества и чистоты АФС, подлежат аттестации.
12.2 Документация по аттестации (испытаниям)
12.20 Для каждого процесса, подлежащего аттестации, следует разработать методику проведения аттестации, согласованную между службой качества и другими отделами, имеющими к ней отношение.
12.21 В методике аттестации должны быть определены критические операции (стадии) технологического процесса и критерии приемлемости, а также виды аттестации (например, ретроспективная, перспективная, текущая) и количество производственных циклов, необходимых для проведения аттестации.
12.22 Отчет о проведении аттестации должен содержать ссылку на методику аттестации (с указанием всех замеченных отклонений) и выводы с рекомендациями по устранению недостатков.
12.23 Любые отклонения от требований методики аттестации должны быть оформлены в письменном виде и утверждены.
12.3 Этапы аттестации (испытаний)
12.30 До проведения работ по аттестации процессов следует провести аттестацию критического оборудования и вспомогательных систем в необходимом объеме. Как правило, такую аттестацию проводят в несколько этапов:
- аттестация проекта (design qualification; DQ) - документальное подтверждение того, что проект производства (в т.ч. помещения, оборудование или вспомогательные системы) соответствует заданию на проектирование и действующим нормам;
- аттестация установленного оборудования (Installation Qualification; IQ) - документальное подтверждение того, что установленное или измененное оборудование, помещения или вспомогательные системы соответствуют утвержденному проекту, заданию на проектирование и действующим нормам;
- аттестация функционирующего оборудования (Operational Qualification; OQ) - документальное подтверждение того, что установленное или измененное оборудование, помещения или вспомогательные системы функционируют надлежащим образом в требуемом режиме;
- аттестация в эксплуатации (Performance Qualification; PQ) - документальное подтверждение того, что оборудование, помещения и вспомогательные системы могут работать совместно с нужной эффективностью и воспроизводимостью в соответствии с утвержденными требованиями и характеристиками технологического процесса.
12.4 Аттестация (испытания) процесса
12.40 Аттестация (испытания) процесса - документальное подтверждение того, что процесс, протекающий в пределах установленных параметров, обеспечивает эффективное и воспроизводимое производство промежуточных продуктов и АФС, удовлетворяющих установленным требованиям и показателям качества.
12.41 Из трех видов аттестации предпочтительным является перспективная аттестация.
12.42 Перспективную аттестацию применяют для всех процессов производства АФС, определенных в 12.12. Перспективная аттестация, используемая для процесса производства АФС, должна быть завершена до выпуска на рынок готового лекарственного средства, произведенного из этой АФС.
12.43 Текущая аттестация может проводиться при отсутствии данных о повторных технологических циклах. Это может быть обусловлено производством только ограниченного числа серий АФС, нерегулярным производством серий АФС или производством АФС с использованием аттестованного процесса, в который были внесены изменения. До завершения текущей аттестации серии АФС могут быть выпущены и использованы для производства готовых лекарственных средств для их реализации на рынке на основании тщательного непрерывного контроля и проведения испытаний этих серий АФС.
12.44 Ретроспективную аттестацию применяют как исключение для хорошо отлаженных процессов, которые не претерпевали значительных изменений для качества АФС из-за изменений сырья, оборудования, систем, устройств или применяемого технологического процесса. Этот вид аттестации может использоваться в том случае, если:
1) определены критические характеристики качества и критические параметры процесса;
2) установлены соответствующие внутрипроизводственные критерии приемлемости и контроля;
3) отсутствовали существенные сбои в ходе процесса (качестве продукции) по причинам, не связанным с ошибками оператора или отказами оборудования;
4) установлен состав примесей для существующих АФС.
12.45 Серии продукции, отобранные для проведения ретроспективной аттестации, должны представлять собой репрезентативную выборку из всех серий, произведенных за рассматриваемый период времени, в т.ч. всех серий, которые не соответствовали требованиям спецификаций. Число этих серий должно быть достаточным для подтверждения стабильности процесса. При проведении ретроспективной аттестации допускается использовать архивные образцы.
12.5 Программа аттестации (испытаний) процесса
12.50 Число производственных циклов для проведения аттестации (испытаний) зависит от сложности процесса или характера изменения параметров процесса. Для проведения перспективной и текущей аттестации следует использовать не менее трех последовательно произведенных серии продукции надлежащего качества. Однако могут быть случаи, при которых для доказательства стабильности процесса могут потребоваться дополнительные производственные циклы (например, сложные или длительные процессы производства АФС). При ретроспективных испытаниях для подтверждения стабильности процесса исследуют данные 10-30 последовательных серий продукции. При наличии обоснования это число может быть уменьшено.
12.51 В ходе аттестации (испытаний) процесса следует контролировать его критические параметры. Параметры процесса, не связанные с качеством продукции (например, расход энергии или срок использования оборудования), допускается не включать в процесс аттестации.
12.52 Аттестация процесса должна подтвердить, что установленный состав примесей АФС находится в допустимых пределах. Состав примесей (качественный и количественный) должен быть аналогичен или лучше состава примесей, установленного по предыдущим данным, или, где применимо, быть лучше состава примесей, установленного при разработке технологического процесса, или профиля примесей в сериях продукции, предназначенных для проведения клинических или токсикологических исследований.
12.6 Периодическая оценка аттестованных (испытанных) систем и процессов
12.60 Системы и процессы следует периодически оценивать для подтверждения того, что они продолжают работать в аттестованном режиме. Повторная аттестация не требуется, если в систему или процесс не было внесено никаких существенных изменений и анализ качества продукции подтверждает, что эта система или процесс стабильно производят продукцию в соответствии с установленными требованиями.
12.7 Аттестация методов очистки
12.70 Как правило, следует проводить аттестацию методов очистки. Обычно аттестацию методов очистки проводят в случаях, когда загрязнение или перенос материалов представляют наибольший риск для качества АФС. Например, на ранних стадиях производства может оказаться ненужной аттестация методов очистки оборудования, если все остатки удаляются последующими операциями очистки.
12.71 Аттестация методов очистки должна отражать фактический характер использования оборудования. Если на одном и том же оборудовании производятся различные АФС или промежуточные продукты, и очистка этого оборудования проводится одним и тем же способом, то для аттестации очистки может быть выбран репрезентативный образец промежуточного продукта или АФС. Этот выбор должен быть основан на свойствах растворимости вещества и трудности его очистки, а также на расчете допустимых предельных значений остатков на основе его активности, токсичности и стабильности.
12.72 Методика аттестации очистки должна содержать характеристику оборудования, подлежащего очистке, порядок очистки, перечень материалов, допустимые предельные значения чистоты, параметры, которые следует контролировать, и применяемые аналитические методы. В методике также следует указать вид отбираемых проб, методы их отбора и маркировки.
12.73 Для определения растворимых и нерастворимых остатков при отборе проб следует использовать такие методы, как взятие мазков, смывов или альтернативные методы (например, прямую экстракцию). Используемые методы отбора проб должны позволять проводить количественное определение остатков на поверхности оборудования после его очистки. Отбор проб путем взятия мазков может быть не всегда применим, если поверхности контакта с продуктом являются труднодоступными из-за особенностей конструкции оборудования и/или ограничений процесса (например, внутренние поверхности шлангов, труб, сосудов, реакторов с маленькими отверстиями) или при работе с токсическими материалами, а также с малогабаритным сложным оборудованием (например, с измельчителями и псевдоожижителями).
12.74 Следует использовать аттестованные аналитические методы, обладающие достаточной чувствительностью для определения допустимого уровня остатков или загрязнений. Для каждого метода следует определить его воспроизводимость. Предельные допустимые количества остатков должны быть реально достижимыми, проверяемыми и основываться на остатке наиболее опасного вещества. Предельные допустимые уровни остатков могут устанавливаться на основе минимальной известной фармакологической, токсикологической или физиологической активности АФС или ее наиболее опасного компонента.
12.75 Для процессов, в которых существует ограничение на общее микробное загрязнение или уровень эндотоксинов в АФС, или других процессов, для которых такое загрязнение может представлять опасность (например, при производстве нестерильных АФС, используемых в производстве стерильных препаратов), аттестация очистки (дезинфекции) оборудования должна включать в себя определение уровня загрязнения по микроорганизмам и эндотоксинам.
12.76 После проведения аттестации методы очистки должны контролироваться с определенной периодичностью для подтверждения того, что они сохраняют эффективность при использовании в текущем производстве. Чистоту оборудования можно контролировать аналитическими методами, а если это допускается, - путем визуального осмотра. Визуальный осмотр позволяет определить большие скопления загрязнений на малых площадях, которые могут оставаться необнаруженными при отборе проб и/или проведении анализов.
12.8 Аттестация аналитических методов
12.80 Следует предусматривать аттестацию аналитических методов, за исключением фармакопейных или других утвержденных методов. В каждом конкретном случае следует подтвердить и оформить документально пригодность каждого применяемого аналитического метода.
12.81 При аттестации аналитических методов следует проверять характеристики, включенные в Руководство ICH по аттестации аналитических методов. Объем исследований, выполняемых при аттестации аналитических методов, должен соответствовать целям анализа и стадии процесса производства АФС.
12.82 Перед началом аттестации аналитических методов следует провести аттестацию соответствующего аналитического оборудования.
12.83 Протоколы всех изменений, вносимых в аттестованный аналитический метод, следует сохранять. В протоколах должны быть указаны причина, из-за которой было внесено изменение, и данные, подтверждающие, что точность и достоверность результатов, полученных после внесения этого изменения, соответствуют характеристикам аттестованного метода.
13.10 На предприятии должна быть введена и документально оформлена система контроля изменений, способных влиять на производство и качество промежуточных продуктов и АФС.
13.11 Документальное оформление, рассмотрение и утверждение изменений в сырье, спецификациях, аналитических методах, помещениях, вспомогательных системах, оборудовании (в т.ч. в компьютерном оборудовании), технологических стадиях, печатных и упаковочных материалах и компьютерных программах должны выполняться по утвержденным инструкциям.
13.12 Все предложения по изменениям, касающимся соблюдения требований настоящего стандарта, должны быть подготовлены, рассмотрены и согласованы с соответствующими подразделениями и службой качества.
13.13 Следует оценить возможное влияние предложенных изменений на качество промежуточных продуктов и АФС. При этом может проводиться классификация изменений для определения объема работ при аттестации. Изменения могут быть классифицированы (например, как незначительные или существенные) в зависимости от их характера и степени влияния, которые эти изменения могут оказать на данный процесс. Следует определить и обосновать, какие дополнительные испытания и аттестационные исследования необходимы для подтверждения возможности внесения изменений в аттестованный процесс.
13.14 При внесении изменений следует принять меры по пересмотру всех документов, на содержание которых влияют эти изменения.
13.15 После внесения изменений следует оценить качество первых серий продукции, произведенных или испытанных с учетом этих изменений.
13.16 Следует оценить возможность влияния критических изменений на установленные сроки годности или дату повторного контроля. При необходимости образцы промежуточных продуктов или АФС, произведенных по модифицированному процессу, могут быть включены в программу ускоренного определения стабильности и/или в программу непрерывного контроля стабильности.
13.17 Все производители готовых лекарственных средств, которым поставляют АФС, должны быть извещены об изменениях установленных методов производства и контроля качества, способных оказать влияние на качество АФС.
14.1 Отклонение
14.10 Следует выполнить маркировку промежуточных продуктов и АФС, не удовлетворяющих требованиям спецификаций, и поместить их на карантинное хранение. Эти промежуточные продукты и АФС могут быть вновь использованы в производстве или направлены на переработку в приведенном ниже порядке. Окончательное решение об использовании (утилизации) отклоненных материалов должно быть оформлено документально.
14.2 Повторная обработка
14.20 Как правило, допускается повторное использование промежуточных продуктов и АФС (в т.ч. материалов, не удовлетворяющих требованиям спецификации) в производстве и их повторная обработка путем повторения стадии кристаллизации или других физических или химических стадий обработки (например, дистилляция, фильтрация, хроматография, измельчение), входящих в технологический процесс. Однако в том случае, если такие операции проводят при производстве большинства серий продукции, они должны быть включены в производство в качестве отдельной стадии принятого технологического процесса.
14.21 Продолжение технологической стадии после того как внутрипроизводственный контроль показал, что эта стадия еще не завершена, рассматривают как часть процесса и не считают повторной обработкой.
14.22 Введение не вступившего в реакцию материала вновь в процесс и повторение химической реакции считают повторной обработкой, если такая операция не является частью технологического процесса. Такой повторной обработке должна предшествовать тщательная оценка, гарантирующая, что качество промежуточных продуктов и АФС не ухудшится вследствие возможного образования побочных продуктов и веществ при дальнейшем протекании реакции.
14.3 Переработка
14.30 Перед принятием решения о переработке серии продукции, не соответствующей установленным требованиям, следует провести расследование причин такого несоответствия.
14.31 Для серий продукции, полученных в результате переработки, следует провести ее оценку, испытания и, при необходимости, - испытания на стабильность с документальным оформлением для подтверждения соответствия ее качества качеству продукции, произведенной по базовой технологии. Для методик переработки может использоваться подход текущей аттестации. Это позволяет отработать методику и определить ожидаемые результаты. Допускается единичный выпуск серии переработанной продукции на основании отчета о ее производстве.
14.32 Следует разработать методики сравнения состава примесей в каждой серии переработанной продукции с составом примесей в сериях продукции, произведенных по базовой технологии. Если для описания характеристик серии переработанной продукции недостаточно обычного набора аналитических методов, следует применять дополнительные методы.
14.4 Регенерация материалов и растворителей
14.40 Допускается регенерация реактивов, промежуточных продуктов или АФС (например, из маточных растворов или фильтратов) при наличии утвержденных методик регенерации и соответствия качества регенерированных материалов установленным требованиям.
14.41 Допускается регенерация и повторное использование растворителей в том или другом процессе при условии контроля процессов их регенерации и подтверждения соответствия растворителей установленным требованиям до их повторного использования или смешивания с другими материалами.
14.42 Допускается смешивание свежеприготовленных и регенерированных растворителей и реактивов, если при испытаниях была установлена возможность их применения во всех технологических процессах, в которых они могут использоваться.
14.43 Использование регенерированных растворителей, маточных растворов и других регенерированных материалов должно быть оформлено документально.
14.5 Возврат
14.50 Возвращенные промежуточные продукты или АФС должны иметь соответствующую маркировку и помещаться на карантинное хранение.
14.51 Если условия, в которых хранились или транспортировались возвращенные промежуточные продукты или АФС до или в процессе их возврата, или состояние их упаковок вызывают сомнения относительно качества этих материалов, то возвращенные промежуточные продукты или АФС должны быть переработаны, повторно обработаны или уничтожены в установленном порядке.
14.52 Следует хранить протоколы о всех возвращенных промежуточных продуктах или АФС. Протокол каждого возврата должен содержать:
- наименование и адрес грузополучателя;
- наименование промежуточного продукта или АФС, номер серии и возвращенное количество;
- причину возврата;
- указание на использование или уничтожение возвращенного промежуточного продукта или АФС.
15.10 Все рекламации по качеству, полученные в устной или письменной форме, должны регистрироваться и расследоваться в соответствии с утвержденной инструкцией.
15.11 Протоколы рассмотрения рекламаций должны включать в себя:
- имя, отчество и фамилию, а также адрес лица, предъявившего рекламацию;
- имя, отчество и фамилию, а также, должность и номер телефона лица, принявшего рекламацию;
- суть рекламации (с указанием наименования и номера серии АФС);
- дату поступления рекламации;
- первоначально принятые меры (с указанием ответственных лиц и даты);
- дальнейшие действия;
- ответ на рекламацию (с указанием даты отправки ответа);
- окончательное решение по серии или партии промежуточного материала или АФС.
15.12 Протоколы рассмотрения рекламаций следует хранить для оценки тенденций, частоты и серьезности рекламаций по данному продукту и принятия дополнительных и срочных мер по устранению недостатков.
15.13 Решение об отзыве следует принимать на основе инструкции, определяющей условия отзыва.
15.14 В инструкции по отзыву продукции должны быть указаны лица, участвующие в анализе поступившей информации, процедуры начала отзыва продукции, круг лиц, которых следует проинформировать об отзыве, и порядок использования (утилизации) отозванного материала.
15.15 В случае серьезной или потенциальной опасности для жизни населения следует проинформировать местные, национальные и/или международные компетентные органы и обратиться к ним с просьбой о содействии.
16.10 Все производители, работающие по контракту (в т.ч. лаборатории), должны выполнять требования настоящего стандарта. Особое внимание должно быть уделено вопросам предотвращения перекрестного загрязнения и обеспечению прослеживаемости.
16.11 Заказчик должен оценить способность подрядчика осуществлять на месте делегированные ему операции в соответствии с требованиями настоящего стандарта.
16.12 Ответственность заказчика и подрядчика за соблюдение требований настоящего стандарта, в т.ч. по обеспечению качества, должна быть определена в контракте (договоре) между ними.
16.13 Договор должен позволять заказчику проводить аудит производства подрядчика на его соответствие настоящему стандарту.
16.14 Если допускается работать по договору субподряда, то подрядчик не должен передавать третьей стороне никакой части работы, порученной ему заказчиком, без предварительного согласования с ним.
16.15 Производственные и лабораторные протоколы должны храниться в местах проведения этого вида деятельности и быть легко доступными.
16.16 Не допускается внесение изменений в технологический процесс, оборудование, спецификации, методы испытаний и др., относящиеся к предмету договора, без согласования с заказчиком.
17.1 Область применения
17.10 Этот раздел относится к любым действиям (за исключением производства), связанным с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС.
17.11 Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны соблюдать требования настоящего стандарта и, в частности, данного руководства.
17.2 Прослеживаемость реализуемых промежуточных продуктов или АФС
17.20 При реализации, хранении, переупаковке и перемаркировке необходимо обеспечить полную прослеживаемость реализуемых промежуточных продуктов или АФС. Для этого следует иметь и хранить следующие документы:
- реквизиты первичного предприятия-производителя;
- адрес первичного предприятия-производителя;
- заказы (договоры) на поставку;
- накладные (транспортные документы);
- документы о получении грузов;
- наименования или обозначения промежуточных продуктов или АФС;
- номер серии продукции, указанной производителем;
- документацию на транспортирование и реализацию;
- все подлинные аналитические паспорта, в т.ч. паспорта первичного предприятия-производителя;
- дату проведения повторного контроля или срок годности.
17.3 Обеспечение качества
17.30 Реализация, хранение, переупаковка и перемаркировка промежуточных продуктов или АФС должны проводиться в соответствии с документально оформленной системой управления качеством (см. 3.2).
17.4 Переупаковка, перемаркировка и обращение с промежуточными продуктами или АФС
17.40 Во избежание перепутывания и ухудшения качества и чистоты промежуточных продуктов или АФС переупаковка, перемаркировка, обращение с промежуточными продуктами или АФС и их контроль должны проводиться в соответствии с требованиями данного стандарта.
17.41 Во избежание прямого и перекрестного загрязнения переупаковка продукции должна проводиться в установленных условиях окружающей среды.
17.5 Стабильность
17.50 Для подтверждения установленных сроков годности или даты повторного контроля исследования стабильности должны проводиться в случаях, если АФС или промежуточный продукт переупаковывают в упаковку другого типа, отличающуюся от используемой производителем.
17.6 Передача информации
17.60 При реализации, хранении, переупаковке и перемаркировке промежуточных продуктов или АФС следует передавать заказчикам всю информацию, получаемую от производителя АФС или промежуточных продуктов, касающуюся качества продукции и решений уполномоченных органов, а также передавать производителям информацию от заказчиков.
17.61 При реализации, хранении, переупаковке и перемаркировке промежуточных продуктов или АФС следует предоставлять потребителю наименование первичного предприятия - производителя АФС или промежуточных продуктов и номера поставляемых серий продукции.
17.62 По требованию уполномоченного органа должна предоставляться информация о первичном предприятии - производителе промежуточных продуктов или АФС. Первичное предприятие-производитель может контактировать с уполномоченным органом непосредственно или через своих представителей.
17.63 Следует соблюдать специальные требования к аналитическим паспортам, приведенные в 11.4.
17.7 Работа с рекламациями и отзывами
17.70 Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны хранить протоколы всех поступивших рекламаций и отзывов в соответствии с требованиями, указанными в разделе 15 настоящего стандарта.
17.71 При необходимости эти лица и организации, а также производитель АФС или промежуточного продукта должны рассмотреть рекламацию для принятия решения о дальнейших действиях, в которых могут участвовать другие потребители (заказчики), получавшие этот АФС или промежуточный продукт, уполномоченный орган или все заинтересованные стороны. Соответствующей стороной должно быть проведено расследование рекламации или отзыва продукции, результаты которого должны быть оформлены документально.
17.72 Если рекламация касается производителя АФС или промежуточного продукта, протокол расследования должен содержать любые данные, полученные от данного производителя АФС или промежуточного продукта (в т.ч. дату и необходимую информацию).
17.8 Работа с возвратами
17.80 Работу с возвратом продукции проводят в соответствии с требованиями 14.52. Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны вести документацию по возвращенным АФС и промежуточным продуктам.
18.1 Общие положения
18.10 В настоящем разделе приведены специальные правила производства и контроля качества АФС или промежуточных продуктов путем культивирования (ферментации) клеток с использованием природных или рекомбинантных организмов, которые не были в должной мере указаны в предыдущих разделах. Этот раздел нельзя отделить от общего содержания настоящего стандарта. Все требования, установленные в настоящем стандарте, остаются в силе. Следует обратить внимание на то, что принципы ферментации для "классических" процессов получения низкомолекулярных соединений и процессов, использующих рекомбинантные и нерекомбинантные организмы для получения белков и/или полипептидов, одинаковы, хотя при этом уровень контроля этих процессов будет различным. В случае, если это имеет значение, в данном разделе указываются эти различия. В целом уровень контроля биотехнологических процессов, используемых для производства белков и полипептидов, должен быть более строгим, чем контроль над классическими процессами ферментации.
18.11 Термин "биотехнологический процесс" относится к использованию клеток или организмов, которые для производства АФС были получены или модифицированы с использованием рекомбинантной ДНК, гибридомной или какой-либо другой технологии. АФС, произведенные биотехнологическими методами, обычно представляют собой высокомолекулярные соединения, такие как белки и полипептиды, для которых в этом разделе приведены специфические требования. С помощью технологии рекомбинантной ДНК могут быть также произведены некоторые АФС с низкими молекулярными массами, например, антибиотики, аминокислоты, витамины и углеводы. Уровень контроля за такими АФС должен соответствовать правилам, применяемым для классической ферментации.
18.12 Термин "классическая ферментация" относится к процессам, использующим для производства АФС микроорганизмы, существующие в природе или модифицированные традиционными методами (радиацией, химическим мутагенезом). АФС, получаемые методом "классической ферментации", обычно являются низкомолекулярными соединениями, такими как антибиотики, аминокислоты, витамины и углеводы.
18.13 Получение АФС и промежуточных продуктов из культуры клеток или методом ферментации включает в себя такие биологические процессы, как культивирование клеток или экстракция и очистка веществ из живых организмов. Следует обратить внимание на то, что могут существовать и дополнительные стадии (например, физико-химическая модификация), которые являются частью технологического процесса. Используемые исходные материалы (питательные среды, компоненты буфера) могут обеспечивать возможность роста микробных загрязнений. В зависимости от природы, метода приготовления и целей использования АФС или промежуточного продукта на определенных стадиях производства может потребоваться проведение контроля микробного, вирусного загрязнения и/или уровня эндотоксинов данной продукции.
18.14 Для обеспечения качества АФС и промежуточных продуктов на всех стадиях производства должен быть установлен надлежащий контроль. Так как настоящий стандарт начинается со стадии культивирования клеток (ферментации), предыдущие стадии (например, поддерживание банка клеток) должны проводиться при наличии надлежащего внутрипроизводственного контроля. Настоящий стандарт описывает процесс культивирования (ферментации) клеток с момента, когда из банка клеток поступает емкость с культурой клеток для использования в производстве.
18.15 Для сведения риска загрязнения к минимуму следует использовать необходимое оборудование и проводить контроль окружающей среды. Критерии приемлемости качества и периодичность проведения контроля окружающей среды зависят от технологической стадии и условий производства (открытая, закрытая или замкнутая система).
18.16 В целом, технологический контроль должен учитывать:
- поддерживание в рабочем состоянии рабочего банка клеток;
- правильную инокуляцию и развитие культуры клеток;
- контроль критических рабочих параметров во время ферментации/культивирования клеток;
- непрерывный контроль процесса роста клеток, их жизнеспособности (для большинства процессов культивирования клеток) и продуктивности;
- методы сбора и очистки, при которых происходит удаление клеток, клеточного дебриса и компонентов питательной среды с одновременной защитой промежуточного продукта или АФС от загрязнения (в частности, микробиологического характера) и потери качества;
- непрерывный контроль микробного загрязнения и, при необходимости, уровня эндотоксинов на определенных стадиях производства и
- вопросы защиты от вирусного загрязнения, описанные в Руководстве ICH Q5A*.
________________
* Руководство ICH Q5A "Качество биотехнологических продуктов. Оценка безопасности вирусного заражения биотехнологических продуктов, произведенных из клеток человеческого и животного происхождения" (Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin).
18.17 При необходимости следует показать, что остатки среды, белки клетки-хозяина, другие примеси и загрязнения, связанные с технологическим процессом, удалены.
18.2 Поддерживание банков клеток и ведение протоколов
18.20 Доступ к банкам клеток должен быть ограничен лицами, имеющими на это полномочия.
18.21 Банки клеток должны храниться в специально разработанных условиях, обеспечивающих поддерживание жизнеспособности и предотвращающих загрязнение клеток.
18.22 Следует вести и сохранять протоколы использования образцов из банков клеток и условия их хранения.
18.23 Банки клеток должны периодически проходить контроль пригодности их использования.
18.24 Более подробная информация по содержанию банков клеток изложена в Руководстве ICH Q5A*.
________________
* Руководство ICH Q5A "Качество биотехнологических продуктов. Выделение и характеристика клеточных субстратов, используемых для производства биотехнологических/биологических продуктов" (Quality of Biotechnological Products: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/ Biological Products).
18.3 Культивирование клеток (ферментация)
18.30 Закрытые системы следует применять, по возможности, в тех случаях, когда необходимо добавлять клеточные субстраты, среды, буфер и газы в асептических условиях. Если первичная инокуляция, последующий перенос или добавки (среды, буфер) проводятся в открытых сосудах, для снижения риска заражения должны быть разработаны и применены соответствующие методы и проведен контроль.
18.31 Если микробное загрязнение может оказать влияние на качество АФС, операции с использованием открытых сосудов следует проводить в биобезопасных шкафах (боксах) или других устройствах, обеспечивающих контроль условий окружающей среды.
18.32 При работе с культурами клеток персонал должен быть одет в специальную одежду и соблюдать специальные меры предосторожности.
18.33 Следует проводить непрерывный контроль критических рабочих параметров (например, температуры, рН, скорости перемешивания, добавления газов, давления), который позволяет гарантировать соответствие условий установленным параметрам процесса. Следует также постоянно контролировать рост клеток, жизнеспособность (для большинства культивируемых клеток) и их продуктивность. В зависимости от типа процесса критические параметры могут изменяться, а в случае классической ферментации некоторые параметры можно не контролировать (например, жизнеспособность клеток).
18.34 Оборудование, используемое для культивирования клеток, после окончания процесса должно быть очищено и стерилизовано. Оборудование для проведения ферментации также должно быть очищено, дезинфицировано или стерилизовано.
18.35 Для предотвращения неблагоприятного влияния на качество АФС следует стерилизовать среды культивирования перед использованием.
18.36 Методики обнаружения загрязнения и принятия соответствующих мер должны быть оформлены документально. К ним относятся методики определения влияния загрязнения на продукцию, методы очистки оборудования и приведение оборудования в состояние готовности для дальнейшего использования. Посторонние микроорганизмы, обнаруженные в процессе ферментации, должны быть идентифицированы, а влияние их на качество продукции должно быть определено. При решении вопроса о судьбе произведенной продукции результаты таких оценок должны приниматься во внимание.
18.37 Протоколы о случаях загрязнения следует сохранять.
18.38 После очистки универсального оборудования между циклами производства различной продукции может потребоваться проведение дополнительных испытаний с целью снижения риска перекрестных загрязнений.
18.4 Сбор, выделение и очистка
18.40 Операции по сбору материала (удалению клеток или клеточных компонентов или сбору клеточных компонентов после разрушения клеток) должны проводиться на оборудовании и в зонах, проект (конструкция) которых должен предусматривать снижение риска загрязнения до минимума.
18.41 Следует разработать методики сбора и очистки, приводящие к удалению или инактивации организмов-продуцентов, клеточного дебриса и компонентов среды (оказывающих при этом минимальное влияние на деградацию, загрязнение и потерю качества) и гарантирующие выход промежуточного продукта и АФС надлежащего качества.
18.42 После использования оборудование должно быть очищено и дезинфицировано в установленном порядке. Производство нескольких последовательных серий промежуточного продукта и АФС без промежуточной очистки оборудования допускается только в том случае, если это не оказывает влияния на их качество.
18.43 При использовании открытых систем очистка должна проводиться в условиях, обеспечивающих сохранение качества продукции.
18.44 Если оборудование используется для производства различных видов продукции, то могут применяться дополнительные виды контроля, такие как использование специальных хроматографических смол или проведение дополнительных испытаний.
18.5 Удаление вирусов (стадии инактивации)
18.50 Более полная информация приведена в Руководстве ICH Q5A*.
________________
* Руководство ICH Q5A "Качество биотехнологических продуктов. Оценка безопасности вирусного заражения биотехнологических продуктов, произведенных из клеток человеческого и животного происхождения" (Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin).
18.51 Стадии удаления и инактивации вирусов являются критическими для некоторых технологических процессов и должны проводиться в рамках параметров, установленных при их аттестации (испытаниях).
18.52 Следует предусмотреть меры предосторожности для предотвращения потенциального заражения вирусами, начиная со стадий до и после удаления (инактивации) вирусов. Открытая обработка должна проводиться в зонах, отделенных от других технологических операций и имеющих отдельную систему вентиляции.
18.53 Как правило, для различных стадий очистки не используют одно и то же оборудование. Если оборудование используют для разных стадий очистки, то перед повторным использованием оно должно быть очищено и дезинфицировано соответствующим образом. Следует предпринимать меры, предотвращающие перенос вирусов с предыдущих стадий (например, через оборудование или окружающую среду).
19.1 Общие положения
19.10 При производстве новых АФС, используемых для проведения клинических исследований, при их разработке могут применяться не все виды контроля, рассмотренные в предыдущих разделах данного стандарта. В настоящем разделе приведены специальные требования к этим АФС.
19.11 Контроль производства АФС, предназначенных для клинических исследований, должен соответствовать той стадии разработки лекарственного средства, в состав которого она входит. Методы производства и контроля качества таких АФС должны быть достаточно гибкими для того, чтобы предусмотреть изменения, вносимые в них по мере получения новых знаний о технологическом процессе и продвижении лекарственного средства с доклинических стадий к клиническим стадиям исследований. Если разработка лекарственного препарата достигает стадии, на которой эта АФС производится для применения в лекарственном препарате, предназначенном для проведения клинических исследований, производители должны гарантировать, что производственные мощности, соответствующие методы производства и контроля обеспечивают необходимое качество этой АФС.
19.2 Качество
19.20 К производству АФС, предназначенных для использования при проведении клинических исследований, должны применяться требования настоящего стандарта с надлежащей процедурой выдачи разрешения на реализацию каждой серии.
19.21 Для выдачи разрешения на реализацию или отклонение каждой серии АФС, предназначенной для использования в клинических исследованиях, должен(ы) быть образован(ы) отдел(ы) контроля качества, независимый(е) от производства.
19.22 Отдельные работы по проведению контроля, обычно выполняемые отделом контроля качества, могут проводиться в других подразделениях.
19.23 Мероприятия по контролю качества должны включать в себя проведение контроля исходных и упаковочных материалов, а также промежуточных продуктов и АФС.
19.24 Следует проводить анализ любых затруднений, связанных с производством и качеством продукции.
19.25 Маркировка АФС, используемых в клинических исследованиях, должна контролироваться установленным образом и содержать информацию о том, что это вещество предназначено для исследовательских целей.
19.3 Оборудование
19.30 При проведении всех стадий клинических разработок (в т.ч. при использовании опытных участков или лабораторий) для производства АФС, предназначенных для использования в клинических исследованиях, должен быть установлен порядок проведения калибровки и очистки оборудования и методики оценки его соответствия своему назначению.
19.31 Порядок эксплуатации оборудования должен гарантировать, что при работе с материалами риск загрязнения и перекрестного загрязнения будет минимальными.
19.4 Контроль исходных материалов
19.40 Исходные материалы, применяемые для производства АФС, предназначенные для использования в клинических исследованиях, должны быть испытаны или должны иметь аналитический паспорт поставщика и проверяться на подлинность. Наличие аналитического паспорта поставщика достаточно для подтверждения качества материала, представляющего опасность.
19.41 В некоторых случаях подтверждение пригодности исходного материала для его использования может быть обосновано не только результатами аналитического контроля, но и упрощенными методами (например, проверкой пригодности).
19.5 Производство
19.50 Процесс производства АФС, предназначенных для использования в клинических исследованиях, должен быть зарегистрирован в лабораторных журналах, протоколах серии или путем использования других средств. Эти документы должны включать в себя информацию об использовании технологических материалов, оборудовании, технологическом процессе и научных наблюдениях.
19.51 Ожидаемые выходы продукции могут различаться более значительно и быть менее определенными, чем ожидаемые выходы продукции процессов, выполняемых в промышленном масштабе. Нет необходимости проводить изучение различия выходов продукции.
19.6 Аттестация (испытания)
19.60 Как правило, не требуется проводить аттестацию (испытания) процесса производства АФС, предназначенных для использования в клинических исследованиях, если производится одна серия АФС или из-за изменений, вносимых в технологический процесс при разработке АФС, повторное изготовление серии является затруднительным или неточным. В этот период разработки качество АФС обеспечивается сочетанием контроля, калибровки (поверки) и аттестации оборудования.
19.61 Аттестацию процесса в соответствии с разделом 12 настоящего стандарта следует проводить для серий АФС, предназначенных для реализации, даже если такие серии производятся на опытном или мелкомасштабном производстве.
19.7 Изменения
19.70 Изменения следует вносить по мере приобретения новых знаний и роста масштаба производства. Каждое изменение технологии производства, спецификаций или методик испытаний должно быть зарегистрировано установленным образом.
19.8 Лабораторный контроль
19.80 Несмотря на то что аналитические методы, используемые для оценки качества серии АФС, предназначенных для клинических исследований, не могут быть аттестованы, они должны быть научно обоснованы.
19.81 Должна быть организована система хранения архивных образцов всех серий АФС, обеспечивающая хранение каждого из архивных образцов в достаточном количестве и надлежащих условиях в течение установленного периода времени после получения разрешения на реализацию, завершения клинического исследования или отзыва заявки на новое лекарственное средство.
19.82 Срок годности и дата повторного контроля, указанные в 11.6 настоящего руководства, применимы к существующим АФС, предназначенным для использования в клинических исследованиях. Для новых АФС, находящихся на ранних стадиях клинических исследований, положения 11.6, как правило, применимы.
19.9 Документация
19.90 Система документации должна гарантировать, что информация, полученная в ходе разработки и производства АФС, предназначенных для использования в клинических исследованиях, будет оформлена документально должным образом и доступна для использования.
19.91 Разработка и применение аналитических методов, используемых для подтверждения выпуска серии АФС, предназначенной для клинических исследований, должны быть оформлены документально.
19.92 Должен быть разработан и внедрен порядок хранения протоколов производства и контроля качества АФС и соответствующей документации. Этот порядок должен обеспечивать хранение протоколов и документов в течение установленного периода времени после получения разрешения на реализацию, завершения клинического исследования или отзыва заявки на новое лекарственное средство.
активная фармацевтическая субстанция; АФС (active pharmaceutical ingredient; API): Любое вещество или смесь веществ, предназначенные для производства лекарственных средств, которые в процессе производства лекарственного средства становятся активным ингредиентом этого лекарственного средства. Такие вещества предназначены для проявления фармакологической активности или другого прямого эффекта при диагностике, лечении, облегчении симптомов или профилактике болезни или для воздействия на структуру или функцию организма.
аттестация (qualification): Документальное подтверждение того, что методика, процесс, оборудование, материал или система соответствуют заданным требованиям и их использование дает ожидаемые результаты.
биологическая нагрузка (bioburden): Уровень и тип (например, патогенных или непатогенных) микроорганизмов, которые могут присутствовать в сырье, исходных материалах АФС, промежуточных продуктах или АФС. Биологическая нагрузка не рассматривается как загрязнение, если ее уровни не превышают установленные предельные значения или не происходит обнаружения определенных патогенных микроорганизмов.
внутрипроизводственный контроль (технологический контроль) (in process control (or process control): Контроль, проводимый в ходе технологического процесса с целью проверки соответствия промежуточного продукта или АФС установленным требованиям, по результатам которого могут корректироваться параметры технологического процесса.
вспомогательные средства (process aids): Материалы (за исключением растворителей), используемые в качестве добавок при производстве промежуточного продукта или АФС, которые сами не участвуют в химической или биологической реакции (например, порошок для фильтрования, активированный уголь и т.д.).
выход ожидаемый (yield, expected): Количество или процент теоретического выхода материала, ожидаемый на любой соответствующей стадии производства, определяемый на основании данных, предварительно полученных при производстве этого материала в лабораторных, опытных или промышленных условиях.
выход теоретический (yield, theoretical): Количество, которое было бы получено на любой соответствующей стадии производства, рассчитанное на основе количества используемого материала в предполагаемых условиях отсутствия любых потерь или ошибок при фактическом производстве.
дата повторного контроля (retest date): Дата прохождения материалом повторной проверки для подтверждения его пригодности для дальнейшего использования.
загрязнение (contamination): Нежелательное внесение примесей химического или микробиологического происхождения или постороннего материала в исходный материал, промежуточный продукт или АФС в ходе производства, отбора проб, упаковки или переупаковки, хранения или транспортирования.
инструкция, методика, процедура (procedure): Документ, содержащий указания по выполнению отдельных видов операций (например, по очистке, переодеванию, контролю окружающей среды, отбору проб, проведению испытаний, эксплуатации оборудования), который прямо или косвенно связан с производством промежуточного продукта или АФС.
испытания (validation): Синоним термина "аттестация".
исходный материал АФС (API starting material): Сырье, промежуточный продукт или АФС, которые используются при производстве АФС и внедряются в структуру АФС в качестве существенного элемента. Исходный материал АФС может быть продуктом (материалом), получаемым от одного или более поставщиков по контракту (коммерческому соглашению), или материалом, производимым на самом предприятии. Как правило, для производства АФС используются исходные материалы с конкретными химическими свойствами и структурой.
калибровка (поверка) (calibration): См. общие термины и определения.
карантин (quarantine): Статус материалов, изолированных физически или иным образом на время до вынесения решения об их выпуске или отклонении.
контроль качества (quality control; QC): Проведение проверок или испытаний на соответствие требованиям спецификаций.
критерии приемлемости (acceptance criteria): Числовые предельные значения, диапазоны или другие критерии, применяемые для приемки результатов испытаний.
критический (critical): Стадии технологического процесса, требования к испытаниям, существенным параметрам или условиям, которые следует задать и контролировать для обеспечения соответствия АФС требованиям спецификации.
лекарственная субстанция (drug substance): Синоним термина "активная фармацевтическая субстанция" (см. стр.172*)
________________
* Страница 172 в бумажном оригинале отсутствует. - Примечание изготовителя базы данных.
лекарственный (медицинский) препарат (drug (medicinal) product): Дозированная форма лекарственного средства в первичной окончательной упаковке, предназначенной для продажи.
материал (material): Сырье (исходные материалы, реактивы, растворители), технологические добавки, промежуточные продукты, АФС, упаковочные и печатные материалы.
маточный раствор (mother liquor): Остаточная жидкость после процессов кристаллизации и выделения.
Примечание - Маточный раствор может содержать непрореагировавшие материалы, промежуточные продукты, АФС и/или примеси в существенном количестве, а также использоваться для дальнейшей обработки.
методика аттестации (испытаний) (validation protocol): Документально оформленный план, устанавливающий порядок аттестации (испытаний) и определяющий критерии приемлемости.
Примечание - Например, методика аттестации (испытаний) технологического процесса, конкретно определяющая технологическое оборудование, критические технологические параметры (рабочие диапазоны), характеристики продукции, отбор проб, данные испытаний, которые должны быть собраны, количество циклов аттестации (испытаний) и приемлемые результаты испытаний.
номер серии (партии) (batch number or lot number): Определенное сочетание цифр, букв и/или символов, обозначающее серию продукции. По номеру серии может быть прослежен весь процесс производства серии АФС и ее реализации.
обеспечение качества (quality assurance; QA): Общая совокупность организационных мероприятий, проводимых с целью обеспечения требуемого качества всех АФС и поддерживания систем качества на должном уровне.
Служба (отдел) качества (quality unit(s): Организационное подразделение, независимое от производства, которое несет ответственность за обеспечение и контроль качества, представляющее собой отделы контроля и обеспечения качества либо одно лицо или группу, в зависимости от размера и структуры организации.
отклонение (deviation): Отступление от утвержденной инструкции или установленного стандарта.
перекрестное загрязнение (cross-contamination): Загрязнение материала или продукта другим материалом или продуктом.
переработка (reworking): Обработка промежуточного продукта или АФС, не удовлетворяющих требованиям стандартов или спецификаций, с помощью одной или нескольких технологических стадий, отличающихся от установленного технологического процесса, с целью получения промежуточного продукта или АФС приемлемого качества (например, перекристаллизация с другим растворителем).
повторная обработка (reprocessing): Повторное введение промежуточного продукта или АФС (в т.ч. не удовлетворяющих требованиям спецификаций) в процесс и повторение стадии кристаллизации или другой соответствующей стадии химической или физической обработки (например, дистилляции, фильтрации, хроматографии, измельчения), которая является частью установленного технологического процесса. Продолжение технологической стадии после того как внутрипроизводственный контроль показал, что она еще не завершена, считается частью текущего процесса производства и не рассматривается как повторная обработка.
подпись (согласование) (signature (signed): См. термин "согласование".
подрядчик (contract manufacturer): Производитель, выполняющий некоторые производственные операции от имени первоначального производителя.
примесь (impurity): Компонент, присутствующий в промежуточном продукте или АФС, который является нежелательным для этого продукта.
производство (manufacture): Операции и виды контроля, связанные с закупкой исходных материалов, их приемкой, обработкой, упаковкой, выпуском в реализацию, хранением и отгрузкой АФС.
промежуточный продукт (intermediate): Материал, получаемый на стадиях производства АФС, который претерпевает дальнейшие молекулярные изменения или очистку до того, как он может считаться АФС. Промежуточные продукты могут не выделяться в качестве чистых продуктов.
Примечание - Настоящий стандарт распространяется только на промежуточные продукты, производство которых начинается со стадии, являющейся, по документам производителя, началом производства АФС.
растворитель (solvent): Неорганическая или органическая жидкость, используемая в качестве носителя для приготовления растворов или суспензий при производстве промежуточного продукта или АФС.
серия (партия) (batch or lot): Определенное количество материалов, произведенных в одном или нескольких технологических процессах так, что их можно считать однородными в рамках установленных пределов. При непрерывном производстве понятие серии может относиться к определенной части продукции, характеризуемой однородностью. Размер серии может быть установлен по фиксированному количеству продукции либо по количеству продукции, производимой за установленный период времени.
система с компьютерным управлением и контролем (computerized system): Система, включающая в себя ввод данных, их электронную обработку и вывод информации, используемой для регистрации или автоматического контроля.
согласование (подпись) (signed signature): Подпись лица, выполняющего конкретное действие или обзор, в виде сокращенной или полной фамилии и инициалов, сделанной от руки, личной печати или авторизованной и защищенной электронной подписи.
состав примесей (impurity profile): Описание идентифицированных и неидентифицированных примесей, присутствующих в АФС.
спецификация (specification): Перечень испытаний, ссылок на аналитические методики и соответствующие критерии приемлемости, устанавливающие числовые границы, диапазоны или другие критерии для указанных испытаний. Спецификация устанавливает критерии, которым должен соответствовать материал для того, чтобы его можно было применять по назначению. Материал соответствует требованиям спецификации, если при испытаниях по принятым аналитическим методам он удовлетворяет установленным критериям приемлемости.
срок годности (expiry date or expiration date): Дата, указываемая на упаковке/этикетке АФС, обозначающая период времени, в течение которого гарантируется сохранение свойств АФС в рамках установленных спецификаций при хранении в определенных условиях и после которого данная АФС не должна использоваться.
сырье (raw material): Исходные материалы, реактивы и растворители, предназначенные для изготовления промежуточного продукта или АФС.
технологический контроль (process control): См. термин "внутрипроизводственный контроль".
технологический процесс (production): Операции, включающие в себя приемку исходных материалов, их обработку, упаковку и получение готовой АФС.
упаковочный материал (packaging material): Любой материал, предназначенный для защиты промежуточного продукта или АФС при хранении и транспортировании.
эталонный стандарт, вторичный (reference standard, secondary): Вещество установленного качества и чистоты по результатам сравнения с первичным эталонным стандартом, используемое в качестве стандарта сравнения для текущих лабораторных анализов.
эталонный стандарт, первичный (reference standard, primary): Вещество, признанное подлинным материалом высокой степени чистоты по результатам обширных аналитических испытаний. Этот стандарт может быть: 1) получен из официально признанного источника; 2) приготовлен путем независимого синтеза; 3) получен из имеющегося промышленного материала высокой степени чистоты или 4) приготовлен путем дальнейшей очистки существующего промышленного материала.